【第119回日本内科学会レポート】糖尿病の病態に関する分子生物学的解析、治療薬のメカニズム――開発が進む新たな治療法とは(8800字)
内分泌系疾患>糖尿病

熊本大学大学院生命科学研究部 代謝内科学 教授
荒木 栄一先生
インスリン発見から約100年が経過し、インスリンの臨床応用やインスリン以外の糖尿病治療薬の開発など、糖尿病診療は大きく進歩した。また、インスリンの生合成・分泌機構や作用機序が分子レベルで解明され、糖尿病の病態や疾患感受性遺伝子の解析も進んできている。
荒木 栄一氏(熊本大学大学院生命科学研究部 代謝内科学 教授)は、第119回日本内科学会総会・講演会(2022年4月15日〜17日)で行われた招聘講演において、糖尿病研究の進歩や糖尿病治療薬の作用機序について解説したうえで、同氏らが取り組んでいる新たな糖尿病治療法の開発状況について紹介した。
インスリンの発見とインスリン療法の始まり
インスリンは1921年に発見され、翌年から1型糖尿病の治療に用いられるようになった。その後1型糖尿病患者の平均余命は著しく改善されたが、糖尿病網膜症・腎症・神経障害といった慢性合併症が新たな問題として浮かび上がった。
そこで、厳格な血糖管理が1型糖尿病の慢性合併症の発症・進展を阻止するのかについて検討すべく行われたのがDCCT(Diabetes control and complications trial)だ。HbA1cは従来療法群(1日1〜2回のインスリン注射)で9%弱、強化療法群(1日3回以上のインスリン注射またはインスリンポンプ療法)では7%強でそれぞれ10年間維持された。その結果、糖尿病網膜症・腎症・神経障害の発症・進展のリスクはいずれも、強化療法群で有意に低下していた。さらに、HbA1cの上昇に応じて慢性合併症の発症リスクが高くなる関係も示され、細小血管障害が血糖値に依存し、発症・進展することが明らかとなった。
2型糖尿病患者において同様の検討を行ったのが、Kumamoto Studyである。強化療法によって良好な血糖コントロールが得られ、慢性合併症の発症・進展リスクも低下する結果が示された。さらに同研究により、HbA1cが6.9%未満、空腹時血糖値が110mg/dL未満、食後2時間血糖値が180mg/dL未満であれば、網膜症・腎症の進行を抑制できる可能性が示唆された。この結果も考慮され、日本糖尿病学会では2013年に熊本宣言として、HbA1c 7.0%未満を合併症予防のための目標値として提唱した。
インスリンの生合成と分泌機構
インスリン遺伝子は、膵β細胞内で転写・翻訳されてプレプロインスリンが合成され、このN末端のシグナルペプチドが切断されてプロインスリンとなる。次に、プロインスリンのA鎖とB鎖との間でジスルフィド結合が形成され、カルボキシペプチダーゼEやプロホルモン転換酵素により切断・分解され、A鎖とB鎖からなるインスリンとCペプチドとが合成され、β顆粒内に貯蔵される。
<インスリンの生合成>
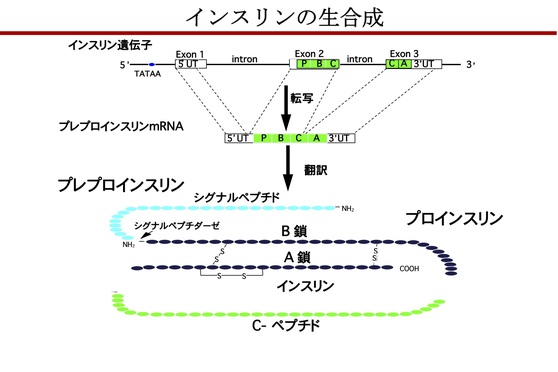
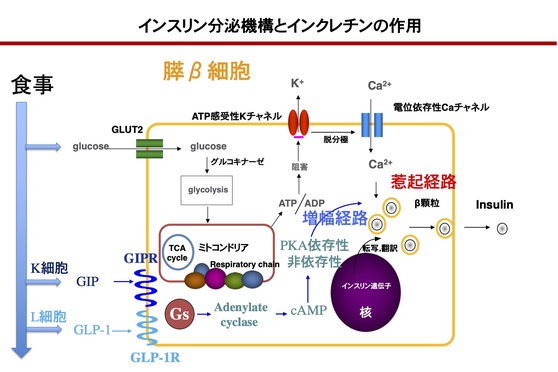
食後に血糖値が上昇すると、GLUT2(glucose transporter type 2)を介してグルコースが膵β細胞内に取り込まれ、クエン酸回路で代謝されてATP(adenosine triphosphate)が産生される。細胞内のATP増加に伴いATP感受性Kチャネルが閉鎖して細胞膜の脱分極が起こることで電位依存性Caチャネルが開口し、細胞外から細胞内にCa2+が流入する。この刺激によりβ顆粒が開口分泌し、血液中にインスリンが分泌される。これが、ブドウ糖反応性のインスリン分泌機序(惹起経路)である。さらに食事の栄養素は、小腸のK細胞、L細胞からインクレチンであるGIP(glucose-dependent insulinotropic polypeptide)とGLP-1(glucagon-like peptide-1)の分泌を促す。インクレチンは、それぞれの受容体を介して細胞内にシグナルを伝え、惹起経路を増幅する機序(増幅経路)としてはたらく。この増幅経路は、血糖値が高いときのみ活性化する。
<インスリン分泌機序とインクレチンの作用>
インスリンの作用機序
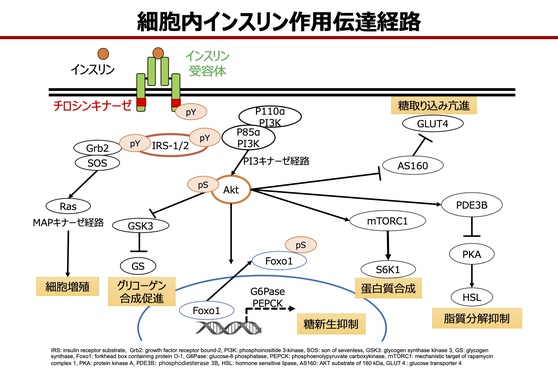
インスリン受容体は、αとβのサブユニットが2つずつ結合したヘテロ4量体構造を持つ。インスリンがαサブユニットに結合すると、細胞内にあるβサブユニットのチロシンキナーゼが活性化され、細胞内基質のIRS(insulin receptor substrate:インスリン受容体基質)-1および2などがチロシンリン酸化を受ける。IRSにはチロシンリン酸化可能部位が散在しており、チロシンリン酸化によって各々にさまざまなシグナル伝達タンパクが結合することで、多様なインスリン作用が発現される。
たとえば、あるチロシンリン酸化部位にはGrb2(growth factor receptor-bound protein2)/SOS(son on sevenless)が結合し、MAP(mitogen-activated protein)キナーゼ経路を活性化してインスリンの増殖作用が伝達される。また、他のチロシンリン酸化部位にはPI-3(phosphatidylinositol 3)キナーゼが結合し、グリコーゲン合成の促進、糖新生の抑制、糖取り込みの亢進といったインスリンの代謝作用が伝達される。これらの機序により、インスリンは種々の標的臓器において、多様な作用を発現して血糖値を調整する。
<細胞内インスリン作用伝達経路>
2型糖尿病の成因
2型糖尿病は、インスリン分泌の障害(インスリン分泌不全)とインスリン作用の障害(インスリン抵抗性)が相まって発症する。インスリン抵抗性は肥満や過栄養、身体運動低下、糖毒性、慢性高インスリン血症などによって引き起こされることが分かっている。
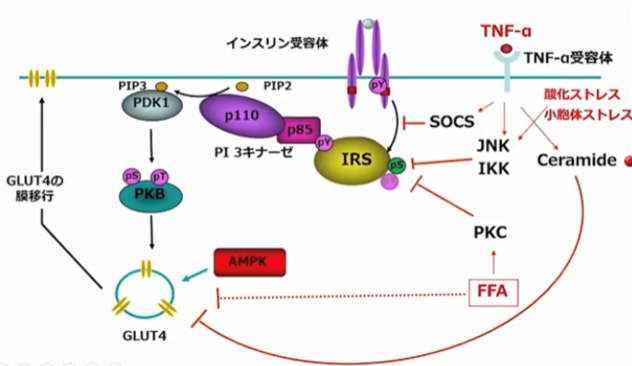
脂肪細胞はさまざまな生理活性物質を分泌する。そのうち、大型の脂肪細胞は遊離脂肪酸やTNFα(tumor necrosis factorα)などを分泌し、これらがインスリン作用を障害してインスリン抵抗性をもたらす。一方で、小型の脂肪細胞からはアディポネクチン分泌が多く、インスリン作用の改善にはたらく。すなわち、小型の脂肪細胞はインスリン感受性を改善に導き、脂肪細胞が肥大化するとインスリン抵抗性が悪化する。
TNF αはインスリン標的臓器でTNF α受容体と結合し、SOCS(suppressor of cytokine signaling)経路、JNK(c-Jun N-terminal kinase)・IKK(inhibitor kappa B kinase)経路、セラミドの活性化を介し、インスリン受容体によるチロシンリン酸化やGLUT4(glucose transporter type4)の細胞膜への移行を抑制する。また、酸化ストレスや小胞体ストレスも、ストレス誘導酵素の代表であるJNKを活性化し、IRS-1のセリンリン酸化を促してインスリン受容体によるチロシンリン酸化を阻害する。脂肪酸の増加に伴い活性化されたプロテインキナーゼCも、IRSやインスリン受容体のセリンリン酸化を促し、同様にインスリン受容体によるチロシンリン酸化を阻害する。このような機序で、インスリン抵抗性が生じる。
<インスリン抵抗性の機序>
膵β細胞においても、酸化ストレスによりJNKが活性化される。その結果、転写因子のFoxO1(Forkhead box transcription factor O1)が核内に移行し、インスリンやグルコキナーゼなどβ細胞機能に重要な遺伝子の発現を調整している転写因子のPdx-1(Pancreas duodenum homeobox 1)が核外に追いやられ、β細胞機能が低下することも分かってきた。
また小胞体ストレス応答経路が、インスリン抵抗性やインスリン分泌不全と関係することも報告されている。細胞小器官の小胞体は、タンパクの合成・品質管理を行う。折りたたみ不全タンパクが小胞体内で増加すると、それを排除するために折りたたみ不全タンパクを正常化する小胞体シャペロンの転写が誘導される。そして、新たなタンパクが合成されないようにmRNAの翻訳が抑制され、折りたたみ不全タンパクを分解する経路(小胞体関連分解)も活性化される。さらに、小胞体ストレスが過剰に加わると、CHOP(C/EBP-homologous protein)という転写因子が誘導され、アポトーシスが起きて細胞全体が排除される。
2型糖尿病患者の膵β細胞では、インスリン抵抗性に伴いインスリン産生が代償性に増加する。その結果、折りたたみの悪いプロインスリンが増加して過剰な小胞体ストレスを生じ、最終的にアポトーシス、β細胞障害に至ることが仮説として考えられている。
2型糖尿病の疾患感受性遺伝子に関する研究も進んでいる。遺伝子変異を全ゲノム領域にわたって関連解析するGWAS(genome wide association study)によって、2型糖尿病の発症リスクを規定する遺伝子が数多く同定されている。なお、これらの遺伝子は単独で糖尿病発症に及ぼす影響は小さく、複数の遺伝子変異が重なることで発症リスクを規定していると考えられている。
実際に、インスリン抵抗性の指標であるHOMA-IR(homeostasis model assessment of insulin resistance)を横軸、インスリン分泌能の指標であるHOMA-β(homeostasis model assessment of βcell)を縦軸とし、2型糖尿病の疾患感受性遺伝子をプロットした研究では、インスリン抵抗性、インスリン分泌不全、あるいはその両方に関係する遺伝子がそれぞれ存在することが示されている。また疾患感受性遺伝子には、民族によらず共通のものと、民族特有のものが認められている。さらに、2型糖尿病の発症リスクを推定するPRS(polygenic risk score)も提唱されている。これは、2型糖尿病の発症と関連する遺伝子型の個数と、それぞれの疾患感受性遺伝子のリスク上昇率(オッズ比)とを掛け合わせて合計し、発症リスクを求めるものである。実際にPRSの上昇に従い、2型糖尿病の有病率が高まること、カットオフ値の設定により高い確率で2型糖尿病の発症リスクを推定できることが報告されている。
2型糖尿病の発症に関与する環境因子では、肥満・過食・高脂肪食・ストレス・加齢といった古典的なものに加え、エピゲノム、母体の肥満、出生時体重、嗜好品、睡眠、運動、腸内細菌なども挙げられている。2型糖尿病の成因である環境因子・遺伝因子を明らかにすることで発症リスクの推定と発症予防法の確立につながり、新たな糖尿病治療標的の同定も期待できる。
糖尿病治療薬の作用機序
2型糖尿病の血糖降下薬は、インスリン分泌非促進系、インスリン分泌促進系、インスリンの3つに分けられる。インスリン分泌非促進系にはビグアナイド薬、チアゾリジン薬、α-グルコシダーゼ阻害薬、SGLT2阻害薬が含まれる。インスリン分泌促進系は血糖依存性と血糖非依存性に分けられ、前者はDPP-4阻害薬とGLP-1受容体作動薬、いわゆるインクレチン関連薬であり、後者にはSU薬と速効型インスリン分泌促進薬(グリニド薬)がある。
ビグアナイド薬「メトホルミン」の作用機序
ビグアナイド薬であるメトホルミンは、肝臓における糖新生抑制、脂肪組織や骨格筋におけるインスリン抵抗性の改善、小腸における糖吸収抑制など多面的な作用機序を持つ。その主要なメカニズムの1つに、AMP(Adenosine monophosphate)キナーゼの活性化がある。
メトホルミンはOCT1(organic cation transporter 1)を介して細胞内に取り込まれ、ミトコンドリアの呼吸鎖Complex Iを阻害する。これによってATPが減少してAMPが増加し、AMPキナーゼが活性化される。その結果、肝臓で糖新生抑制、脂肪酸β酸化の亢進、脂肪酸の合成抑制が生じる。一方でAMPキナーゼ活性以外の経路もいくつか報告されている。たとえば、肝細胞におけるAMPの増加がアデニル酸シクラーゼ活性を低下させ、グルカゴンのシグナルを抑制する。その結果、グルカゴンによる糖新生系の酵素の発現を抑え、AMPキナーゼとは独立した機序で、肝臓における糖新生を抑制するというものだ。
そのほか、メトホルミンによる小腸からのシグナルが、延髄孤束核を経て迷走神経遠心線維を活性化し、肝臓での糖産生を抑制するという報告や、低用量のメトホルミンがライソゾームのプロトンポンプであるvATPase(vacuolar adenosine triphosphatase)活性を阻害することで、ミトコンドリアを介さずにAMPキナーゼを活性化するという報告が、それぞれ基礎研究の結果として示されている。
チアゾリジン誘導体の作用機序
チアゾリジン誘導体の標的分子はPPARγ(Peroxisome Proliferator-Activated Receptor γ)である。チアゾリジン誘導体がPPARγに結合すると、特定のDNAに対する転写活性が上昇し、種々の遺伝子発現が誘導される。その結果、大型の脂肪細胞が減少し、小型の脂肪細胞が増加することでアディポサイトカインが変化する。また脂肪酸代謝に関与する分子にも変化が起こる。このような機序でインスリン抵抗性が改善すると予想されている。
αグルコシダーゼ阻害薬、SGLT2阻害薬の作用機序
αグルコシダーゼ阻害薬は、小腸において二糖類から単糖類への分解を阻害し、糖吸収を緩やかにして食後の血糖上昇をなだらかにする。SGLT2阻害薬の作用部位は腎尿細管である。通常、糸球体でろ過されたグルコースのうち、近位尿細管のSGLT2で約90%、SGLT1で約10%が再吸収される。SGLT2阻害薬は糖再吸収を阻害して尿糖を増やし、血糖値を下げる効果がある。
インスリン分泌促進系薬の作用機序
SU薬、グリニド薬は、膵β細胞においてATP感受性Kチャネルに結合し、血糖非依存性に惹起経路を活性化してインスリンを分泌させる。一方でインクレチン関連薬のDPP-4阻害薬とGLP-1受容体作動薬は、増幅経路を活性化して血糖依存性にインスリンを分泌させる作用を持つ。
新たな糖尿病治療薬であるイメグリミンは、メトホルミンと構造式が類似している。作用機序もメトホルミンと同様、ミトコンドリアの呼吸鎖Complex Iを阻害し、肝臓での糖新生を抑制することでインスリン抵抗性を改善させる。それに加え、膵β細胞において細胞内カルシウムイオン濃度を増加させ、ブドウ糖依存性のインスリン分泌促進作用も有している。
糖尿病の成因解明や治療法開発に関する取り組み
最後に、生活習慣への集団介入(田原坂スタディ)、褐色脂肪細胞の活性化、ミトコンドリア由来活性酸素の制御、Heat shock protein 72(HSP72)の誘導に関する当科の研究成果を紹介する。
田原坂スタディ――メタボリックシンドローム予備軍に対する生活習慣介入試験
田原坂スタディは、熊本県植木町の住民における生活習慣病改善のための集団介入の有効性を検討した研究である。全人口約3万人(2002年当時)のうち2,986人の健診データを解析し、耐糖能異常・脂質異常症・高血圧・肥満のいずれか1つ以上を有した417名をピックアップし、同意の得られた137名を対象とした。
この対象者を、まったく介入しないコントロール群、月に1回ずつ4か月間講義中心の介入を行った標準介入群、標準介入後に月に1回ずつ集団運動指導を行った延長介入群の3つに分け、介入終了時(10か月後)、介入終了1年後(22か月後)、介入終了2年後(34か月後)まで追跡した。
その結果、BMIはコントロール群では変化がなかったが、標準介入群および延長介入群では有意な減少を認め、介入終了2年後まで維持されていた。さらにメタボリックシンドロームを構成する因子の数も、コントロール群では経時的に増加する一方、標準介入群と延長介入群では因子数の増加が抑制されていた。これらの結果より、専門家による集団への介入がメタボリックシンドローム危険因子を改善し、その効果が少なくとも2年間は持続することが示された。
褐色脂肪細胞の活性化
脂肪細胞には白色脂肪細胞と褐色脂肪細胞があり、カロリー消費を行う褐色脂肪細胞の活性化により、代謝が高まり体重が減少することが期待される。脂肪細胞でのインスリン作用を特異的に阻害したマウスとコントロールのマウスの血清タンパクを比較し、褐色脂肪細胞を活性化する因子を同定してBAF(Brown adipocyte activation factor)と名付けた。
今後、BAFが褐色脂肪細胞を活性化するメカニズムや、BAFと白色脂肪細胞のベージュ化(褐色脂肪細胞と同様に熱産生を行うベージュ脂肪細胞への移行)の関連について検討することで、肥満や糖尿病の治療薬開発に役立つことが期待される。
ミトコンドリア由来活性酸素の制御
高血糖により増加したミトコンドリア由来活性酸素種は、糖尿病合併症の成因の1つであることが提唱されている。ミトコンドリア由来活性酸素種のマーカーである尿中8-OHdGは、日本人2型糖尿病患者においてHbA1cと有意に正相関し、網膜症・腎症・大血管障害の合併により上昇していると報告している。
そこで、ミトコンドリア由来活性酸素を除去する酵素であるMnSOD(Manganese superoxide dismutase)を血管内皮に発現させた糖尿病マウスにおいて、合併症に関する検証を行った。すると、糖尿病を誘発しても尿中8-OHdGが上昇せず、糖尿病網膜症に関与するVEGF(vascular endothelial growth factor)の発現が抑制され、血管新生を生じづらいことが分かった。さらに、メトホルミンやピオグリタゾンは、遺伝子転写を制御するPGC-1αを活性化し、MnSODやミトコンドリア数を増加させることが明らかになった。これらの糖尿病治療薬は血糖降下作用に加えて、ミトコンドリア由来活性酸素種を減少させ、糖尿病合併症を抑制することが予測された。
Heat shock protein 72(HSP72)の誘導
熱によって発現誘導されるシャペロンのHSP72は、ストレス誘導酵素であるJNKの活性を抑制する作用を持つ。我々は温熱と同時にパルス状直流電流を加えるMET療法により、HSP72が強く誘導されることを明らかにし、糖尿病治療への活用について検討している。
たとえば、糖尿病モデルの高脂肪食負荷マウスでは、MET療法により空腹時・糖負荷後血糖値の低下、インスリン感受性の改善、内臓脂肪の減少、肥満に伴うアディポネクチンの減少抑制を認めている。また、肝臓ではPI-3キナーゼ活性が高まり、インスリン受容体のリン酸化が増えて、下流のシグナルが回復していることも示された。
また、健常人において安全性を確認した後、肥満の2型糖尿病患者においてもMET療法を行ったところ、内臓脂肪の減少、血圧低下、インスリン抵抗性の改善、高感度CRPなど慢性炎症を示唆するパラメーターの低下、アディポネクチンの上昇が認められた。さらにMET療法の頻度を増やすと、回数依存性に内臓脂肪面積が減少し、HbA1cが低下する結果も得られた。
MET療法前後での末梢血の単球を解析した結果、ストレス誘導酵素の発現を活性化する転写因子のNF-κBの減少とHSP72の増加、またAMPキナーゼは活性化し、JNK活性は低下していることが確認された。さらに炎症性マーカーのmRNAも減少していた。健常男性やメタボリックシンドロームを有する男性においても、同様の結果が得られている。以上より、MET療法は内臓脂肪を減少させてインスリン抵抗性を改善させる効果、抗炎症効果、血糖改善効果を有することが期待される。
講演のまとめ
- インスリンの分泌機構には惹起経路と増幅経路がある
- SU薬とグリニド薬は血糖非依存性に惹起経路を活性化してインスリンを分泌させ、インクレチン関連薬のDPP-4阻害薬とGLP-1受容体作動薬は、増幅経路を活性化して血糖依存性にインスリンを分泌させる
- インスリン作用は、受容体のチロシンキナーゼが活性化し、細胞内基質がチロシンリン酸化することで伝達される。このプロセスがTNF-α・酸化ストレス・小胞体ストレスなどにより阻害されると、インスリン抵抗性を生じる
- ビグアナイド薬であるメトホルミンの作用には、肝臓における糖新生抑制、脂肪組織や骨格筋におけるインスリン抵抗性の改善、小腸における糖吸収抑制などがある。その主要なメカニズムとして、AMPキナーゼの活性化が挙げられる
- チアゾリジン薬の誘導体は、PPARγに結合すると、大型の脂肪細胞が減少して小型の脂肪細胞が増加し、アディポサイトカインが変化してインスリン抵抗性が改善する
- αグルコシダーゼ阻害薬は、小腸からの糖吸収を緩やかにする
- SGLT2阻害薬は尿細管からの糖再吸収を阻害して尿糖を増やし、血糖値を下げる
- イメグリミンはメトホルミンと同様に、肝糖新生抑制作用によるインスリン抵抗性改善効果を有するほか、膵β細胞におけるブドウ糖依存性のインスリン分泌促進作用も持つ
- 生活習慣への集団介入に加え、褐色脂肪細胞の活性化、ミトコンドリア由来活性酸素の制御、HSP72の誘導など、糖尿病や合併症の成因解明や治療法開発を進めており、これらの臨床応用が期待される
医師について
新着記事
乳がん診療の将来見据え課題解決に注力―日本乳癌学会・戸井雅和理事長インタビュー
乳がん診療の将来見据え課題解決に注力―日本乳癌学会・戸井雅和理事長インタビュー
腫瘍(オンコロジー)>乳腺・乳房
がん・感染症センター 都立駒込病院 院長
戸井雅和先生 医師・歯科医師限定
医師・歯科医師限定超高齢社会見据え運動器寄りにバランスも―第68回日本リウマチ学会総会・学術集会、髙木理彰会長に聞く見どころ・聴きどころ
超高齢社会見据え運動器寄りにバランスも―第68回日本リウマチ学会総会・学術集会、髙木理彰会長に聞く見どころ・聴きどころ
筋骨格系疾患>関節
免疫系疾患>自己免疫疾患
山形大学医学部整形外科学講座 教授
髙木 理彰先生医師・歯科医師限定若手が「発表」で競い合う「ことはじめ甲子園」など新企画も盛りだくさん―第64回日本呼吸器学会学術講演会、4月5日から横浜市で開催
若手が「発表」で競い合う「ことはじめ甲子園」など新企画も盛りだくさん―第64回日本呼吸器学会学術講演会、4月5日から横浜市で開催
呼吸器疾患
腫瘍(オンコロジー)>呼吸器
その他>その他
横浜市立大学大学院医学研究科呼吸器病学教室 主任教授/附属病院副院長
金子 猛先生医師・歯科医師限定日耳鼻・村上理事長に聞く 啓発活動の意味と意義、耳鼻咽喉科領域の課題
日耳鼻・村上理事長に聞く 啓発活動の意味と意義、耳鼻咽喉科領域の課題
耳鼻咽喉疾患
日本耳鼻咽喉科頭頸部外科学会 理事長/名古屋市立大学医学部附属 東部医療センター 特任教授・高次ウイルス感染症センター長
村上 信五先生医師・歯科医師限定内臓脂肪型肥満の発症・進展に白血球中のタンパクが関与―横浜市大の研究グループが解明、医学誌で発表
内臓脂肪型肥満の発症・進展に白血球中のタンパクが関与―横浜市大の研究グループが解明、医学誌で発表
内分泌系疾患>その他
MedicalNoteExpert編集部医師・歯科医師限定「多職種で支える周産期リエゾンのバトン」テーマに日本周産期メンタルヘルス学会学術集会開催――大会長、竹内 崇氏が注目演題など紹介
「多職種で支える周産期リエゾンのバトン」テーマに日本周産期メンタルヘルス学会学術集会開催――大会長、竹内 崇氏が注目演題など紹介
精神疾患
その他>周産期
東京医科歯科大学 大学院医歯学総合研究科 精神行動医科学分野 リエゾン精神医学・精神腫瘍学担当 准教授
竹内 崇先生医師・歯科医師限定4年ぶり完全オフライン開催――日本血液学会学術集会・豊嶋会長が感じた熱気と開催にかける思い
4年ぶり完全オフライン開催――日本血液学会学術集会・豊嶋会長が感じた熱気と開催にかける思い
腫瘍(オンコロジー)>造血器
血液・リンパ疾患>血液
血液・リンパ疾患>リンパ
北海道大学大学院 医学研究院 内科系部門 内科学分野 血液内科学教室 教授
豊嶋 崇徳先生医師・歯科医師限定実は身近な「医師会」――現場の医師・国民を支える思いと活動、理事2氏が語る
実は身近な「医師会」――現場の医師・国民を支える思いと活動、理事2氏が語る
その他>プライマリケア
その他>その他
小磯診療所院長/神奈川県医師会理事
磯崎 哲男先生医師・歯科医師限定“口腔の番人”口腔外科医……池邉 哲郎理事長に聞く口腔外科学会の課題と展望
“口腔の番人”口腔外科医……池邉 哲郎理事長に聞く口腔外科学会の課題と展望
腫瘍(オンコロジー)>頭頸部
その他>その他
福岡歯科大学 口腔・顎顔面外科学講座 口腔外科学分野 教授
池邉 哲郎先生医師・歯科医師限定実は身近な小児希少難病、専門医へのアクセス改善へ――健やか親子支援協会の取り組み
実は身近な小児希少難病、専門医へのアクセス改善へ――健やか親子支援協会の取り組み
先天性・遺伝性疾患
小児疾患
藤田医科大学 がん医療研究センター センター長/慶應義塾大学 医学部 名誉教授
佐谷 秀行先生医師・歯科医師限定日本神経免疫学会学術集会の見どころ紹介――新薬登場で医師に求められるスキル、会長など4氏に聞く
日本神経免疫学会学術集会の見どころ紹介――新薬登場で医師に求められるスキル、会長など4氏に聞く
脳神経系疾患>その他
免疫系疾患>その他
東京医科歯科大学大学院 医歯学総合研究科 脳神経病態学分野 教授
横田 隆徳先生医師・歯科医師限定補体学会・井上会長に聞く研究の現状―希少疾患の解明・治療の可能性広がるも検査体制、研究者減が課題 啓発・教育に注力し裾野拡大目指す
補体学会・井上会長に聞く研究の現状―希少疾患の解明・治療の可能性広がるも検査体制、研究者減が課題 啓発・教育に注力し裾野拡大目指す
先天性・遺伝性疾患
その他>その他
和歌山県立医科大学分子遺伝学講座 教授
井上 徳光先生医師・歯科医師限定J-OSLERの登録1.4か月分の勤務時間に相当、北大が実態調査――危惧される地方の内科医減少
J-OSLERの登録1.4か月分の勤務時間に相当、北大が実態調査――危惧される地方の内科医減少
その他>その他
北海道大学病院 臨床研修センター副センター長/血液内科 講師
小野澤 真弘先生医師・歯科医師限定褐色脂肪細胞の鍵因子「NFIA」高発現で肥満、糖尿病改善―エネルギー消費促す経口薬の可能性に期待
褐色脂肪細胞の鍵因子「NFIA」高発現で肥満、糖尿病改善―エネルギー消費促す経口薬の可能性に期待
内分泌系疾患>糖尿病
MedicalNoteExpert編集部医師・歯科医師限定日本ではなぜ医療機器の開発が進まないのか? 求められる“ロボット医療”の時代に向けた人材育成――藤澤正人学長に聞く神戸大学の挑戦
日本ではなぜ医療機器の開発が進まないのか? 求められる“ロボット医療”の時代に向けた人材育成――藤澤正人学長に聞く神戸大学の挑戦
その他>その他
神戸大学 学長
藤澤 正人先生医師・歯科医師限定日本産科婦人科学会前理事長・木村 正氏に聞く、産婦人科の魅力や課題――「働き方改革」で周産期医療体制はどうなる
日本産科婦人科学会前理事長・木村 正氏に聞く、産婦人科の魅力や課題――「働き方改革」で周産期医療体制はどうなる
腫瘍(オンコロジー)>その他
感染症>ウイルス性
その他>周産期
大阪大学大学院医学系研究科・医学部 器官制御外科学講座 産科学婦人科学 教授
木村 正先生医師・歯科医師限定学会の存在意義は「研究による社会貢献」―小児神経学会・加藤理事長に聞く小児神経学の魅力、やりがい、課題
学会の存在意義は「研究による社会貢献」―小児神経学会・加藤理事長に聞く小児神経学の魅力、やりがい、課題
小児疾患
脳神経系疾患>その他
昭和大学医学部小児科学講座 教授/昭和大学病院 てんかん診療センター センター長
加藤 光広先生医師・歯科医師限定2型糖尿病は治らない? 実は100人に1人が寛解―新潟大の研究で判明したその条件とは
2型糖尿病は治らない? 実は100人に1人が寛解―新潟大の研究で判明したその条件とは
内分泌系疾患>糖尿病
MedicalNoteExpert編集部医師・歯科医師限定デジタル禁煙療法の有効性は――日本癌学会 がんを始めとする喫煙の健康に関する注目すべき文献紹介
デジタル禁煙療法の有効性は――日本癌学会 がんを始めとする喫煙の健康に関する注目すべき文献紹介
その他>プライマリケア
その他>その他
MedicalNoteExpert編集部医師・歯科医師限定クラファンで若手がん研究者を支援――日本癌学会、秋の学術総会で応援イベント開催
クラファンで若手がん研究者を支援――日本癌学会、秋の学術総会で応援イベント開催
その他>その他
藤田医科大学医科学研究センター 研究員/慶應義塾大学医学部 先端医科学研究所 訪問研究員
大槻 雄士先生医師・歯科医師限定COVID-19によるがん薬物療法への影響――日本臨床腫瘍学会が第7波における実態調査の結果を報告
COVID-19によるがん薬物療法への影響――日本臨床腫瘍学会が第7波における実態調査の結果を報告
腫瘍(オンコロジー)>その他
感染症>ウイルス性
その他>緩和ケア
MedicalNoteExpert編集部医師・歯科医師限定乳歯早期脱落の陰に低ホスファターゼ症の可能性――「遭遇した歯科医は小児歯科専門医と連携を」早期発見に向け呼びかけ
乳歯早期脱落の陰に低ホスファターゼ症の可能性――「遭遇した歯科医は小児歯科専門医と連携を」早期発見に向け呼びかけ
先天性・遺伝性疾患
小児疾患
筋骨格系疾患>骨
その他>プライマリケア
大阪大学大学院歯学研究科 小児歯科学講座 教授/大阪大学歯学部附属病院 小児歯科 科長
仲野 和彦先生医師・歯科医師限定日本耳鼻咽喉科頭頸部外科学会・5月中旬に福岡で学術講演会を開催――会長に聞く、注目演題と開催への思い
日本耳鼻咽喉科頭頸部外科学会・5月中旬に福岡で学術講演会を開催――会長に聞く、注目演題と開催への思い
耳鼻咽喉疾患
腫瘍(オンコロジー)>頭頸部
その他>その他
九州大学大学院医学研究院 耳鼻咽喉科学分野 教授
中川 尚志先生医師・歯科医師限定新薬登場で変わる糖尿病治療――変革期に開く糖尿病学会学術集会の見どころを西尾 善彦会長に聞く
新薬登場で変わる糖尿病治療――変革期に開く糖尿病学会学術集会の見どころを西尾 善彦会長に聞く
内分泌系疾患>糖尿病
その他>その他
鹿児島大学大学院医歯学総合研究科 糖尿病・内分泌内科学 教授
西尾 善彦先生医師・歯科医師限定「究める-知・仁・術-」テーマに日本整形外科学会学術総会・横浜で開催――総会会長に聞く、整形外科の魅力と総会への思い
「究める-知・仁・術-」テーマに日本整形外科学会学術総会・横浜で開催――総会会長に聞く、整形外科の魅力と総会への思い
その他>その他
岡山大学学術研究院医歯薬学域 生体機能再生・再建学講座(整形外科学)教授
尾﨑 敏文先生医師・歯科医師限定「慈心妙手」の精神伝える第75回日本産科婦人科学会の見どころ――6か国の若手医師が議論する場も
「慈心妙手」の精神伝える第75回日本産科婦人科学会の見どころ――6か国の若手医師が議論する場も
その他>その他
東京慈恵会医科大学 産婦人科学講座 主任教授
岡本 愛光先生医師・歯科医師限定「知の融合で拓く未来」見据え日本呼吸器学会学術講演会開催―大会長、髙橋 和久・順天堂大教授が注目演題など紹介
「知の融合で拓く未来」見据え日本呼吸器学会学術講演会開催―大会長、髙橋 和久・順天堂大教授が注目演題など紹介
呼吸器疾患
腫瘍(オンコロジー)>呼吸器
免疫系疾患>自己免疫疾患
感染症>ウイルス性
その他>その他
順天堂大学医学部附属順天堂医院 院長(呼吸器内科 教授)
髙橋 和久先生医師・歯科医師限定BMPR2変異によるHPAHの予後――早期発見を目指した運動負荷検査の取り組み
BMPR2変異によるHPAHの予後――早期発見を目指した運動負荷検査の取り組み
心臓血管疾患>血管
その他>検査
慶應義塾大学医学部 内科学教室循環器内科 難治性循環器疾患病態学寄付研究講座 特任助教
平出 貴裕先生医師・歯科医師限定「外科医の仕事は尊く、素晴らしい」――大木隆生先生に聞く外科医療を取り巻く現状と思い
「外科医の仕事は尊く、素晴らしい」――大木隆生先生に聞く外科医療を取り巻く現状と思い
その他>その他
東京慈恵会医科大学 外科学講座 チェアマン(統括責任者)
大木 隆生先生医師・歯科医師限定慈恵医大外科301人でもてなす第123回日本外科学会学術集会――外科医がより輝くための学術集会に
慈恵医大外科301人でもてなす第123回日本外科学会学術集会――外科医がより輝くための学術集会に
その他>その他
東京慈恵会医科大学 外科学講座 チェアマン(統括責任者)
大木 隆生先生医師・歯科医師限定国内外のエキスパートが集結・日本リウマチ学会4月末に福岡で現地開催――国内外から約2000演題が集まる
国内外のエキスパートが集結・日本リウマチ学会4月末に福岡で現地開催――国内外から約2000演題が集まる
免疫系疾患>自己免疫疾患
その他>その他
産業医科大学医学部 第1内科学講座 教授
田中 良哉先生医師・歯科医師限定糖尿病治療への再生医療応用――β細胞新生・再生研究の現状と展望
糖尿病治療への再生医療応用――β細胞新生・再生研究の現状と展望
内分泌系疾患>糖尿病
北里大学病院 内分泌代謝内科 科長/主任教授
宮塚 健先生医師・歯科医師限定糖尿病診療は双方向のコミュニケーションが重要――医師だけでなくスタッフも参加、患者がアクセスできる窓口を多様化
糖尿病診療は双方向のコミュニケーションが重要――医師だけでなくスタッフも参加、患者がアクセスできる窓口を多様化
内分泌系疾患>糖尿病
北里大学病院 内分泌代謝内科 科長/主任教授
宮塚 健先生医師・歯科医師限定糖尿病治療で求められる地域連携の在り方とは――エリアにより異なる課題も
糖尿病治療で求められる地域連携の在り方とは――エリアにより異なる課題も
内分泌系疾患>糖尿病
北里大学病院 内分泌代謝内科 科長/主任教授
宮塚 健先生医師・歯科医師限定内分泌代謝内科を志したきっかけとやりがい――ワークライフバランスを考えたキャリア形成が可能
内分泌代謝内科を志したきっかけとやりがい――ワークライフバランスを考えたキャリア形成が可能
内分泌系疾患>糖尿病
その他>その他
北里大学病院 内分泌代謝内科 科長/主任教授
宮塚 健先生医師・歯科医師限定「新しい医学を協創する内科学」テーマに日本内科学会総会・講演会開催――AIがもたらす可能性、内科の醍醐味を語る
「新しい医学を協創する内科学」テーマに日本内科学会総会・講演会開催――AIがもたらす可能性、内科の醍醐味を語る
心臓血管疾患>血管
心臓血管疾患>心臓
その他>その他
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定日本小児科学会・4月中旬に東京で学術集会を開催――小児医療の課題とトピックとともに学術集会の見どころを紹介
日本小児科学会・4月中旬に東京で学術集会を開催――小児医療の課題とトピックとともに学術集会の見どころを紹介
小児疾患
その他>その他
順天堂大学大学院医学研究科 小児思春期発達・病態学講座 主任教授
清水 俊明先生医師・歯科医師限定“赤字手術”の点数引き上げ、ロボット支援手術の術者要件廃止……外保連の取り組みと成果――光熱費高騰による病院経営難も
“赤字手術”の点数引き上げ、ロボット支援手術の術者要件廃止……外保連の取り組みと成果――光熱費高騰による病院経営難も
腫瘍(オンコロジー)>消化器
その他>その他
地方独立行政法人埼玉県立病院機構 理事長
岩中 督先生医師・歯科医師限定総会長 相原一・東京大教授に聞く第127回眼科学会総会のみどころ―養老孟司氏講演など「横断的企画」も
総会長 相原一・東京大教授に聞く第127回眼科学会総会のみどころ―養老孟司氏講演など「横断的企画」も
その他>その他
東京大学医学部眼科学教室教授
相原 一先生医師・歯科医師限定【インタビュー】4年ぶり現地開催の再生医療学会 「双方向的議論」の場も―髙橋淳・総会会長が見どころなど紹介
【インタビュー】4年ぶり現地開催の再生医療学会 「双方向的議論」の場も―髙橋淳・総会会長が見どころなど紹介
その他>その他
京都大学 iPS細胞研究所長 教授
髙橋 淳先生医師・歯科医師限定【第71回日本アレルギー学会レポート】PGAM2022改訂のポイント――診断と治療、ステップダウンまで(4000字)
【第71回日本アレルギー学会レポート】PGAM2022改訂のポイント――診断と治療、ステップダウンまで(4000字)
呼吸器疾患
昭和大学医学部 内科学講座 呼吸器・アレルギー内科学部門 主任教授/昭和大学病院 病院長
相良 博典先生医師・歯科医師限定【インタビュー】4年に1度の日本医学会総会――「ビッグデータ」テーマに技術革新がもたらす医学・医療の未来を考える(2100字)
【インタビュー】4年に1度の日本医学会総会――「ビッグデータ」テーマに技術革新がもたらす医学・医療の未来を考える(2100字)
その他>その他
朝日生命成人病研究所 所長/国立国際医療研究センター 名誉理事長
春日 雅人先生医師・歯科医師限定【インタビュー】「Cancer, Science and Life」テーマに20年を振り返る日本臨床腫瘍学会・福岡で3月中旬に開催
【インタビュー】「Cancer, Science and Life」テーマに20年を振り返る日本臨床腫瘍学会・福岡で3月中旬に開催
腫瘍(オンコロジー)>その他
九州大学大学院医学研究院 社会環境医学講座 連携社会医学分野 教授
馬場 英司先生医師・歯科医師限定【第55回日本てんかん学会学術集会レポート】 東日本大震災やCOVID-19パンデミックなどのクライシスに対する医療マネジメント活動――本部の機能と情報収集の重要性
【第55回日本てんかん学会学術集会レポート】 東日本大震災やCOVID-19パンデミックなどのクライシスに対する医療マネジメント活動――本部の機能と情報収集の重要性
感染症>ウイルス性
その他>その他
東北大学病院 総合地域医療教育支援部 教授
石井 正先生医師・歯科医師限定【インタビュー】進化するテクノロジーを駆使し循環器領域の発展に向けたチャレンジを――次世代を担う若者に向けて筒井 裕之氏(九州大学)からのメッセージ
【インタビュー】進化するテクノロジーを駆使し循環器領域の発展に向けたチャレンジを――次世代を担う若者に向けて筒井 裕之氏(九州大学)からのメッセージ
心臓血管疾患>血管
心臓血管疾患>心臓
九州大学大学院医学研究院 循環器内科学 教授
筒井 裕之先生医師・歯科医師限定【インタビュー】日本循環器学会学術集会・福岡で3年ぶり現地開催――対面での活発な交流・議論を期待
【インタビュー】日本循環器学会学術集会・福岡で3年ぶり現地開催――対面での活発な交流・議論を期待
心臓血管疾患>血管
心臓血管疾患>心臓
九州大学大学院医学研究院 循環器内科学 教授
筒井 裕之先生医師・歯科医師限定【第81回日本癌学会レポート】 過去と現在のがん研究――基礎研究者の立場から捉える魅力と意義(4000字)
【第81回日本癌学会レポート】 過去と現在のがん研究――基礎研究者の立場から捉える魅力と意義(4000字)
腫瘍(オンコロジー)>乳腺・乳房
その他>検査
金沢大学がん進展制御研究所 分子病態研究分野 教授
後藤 典子先生医師・歯科医師限定【第84回日本血液学会学術集会レポート】次世代型CAR-T細胞療法の開発動向――遺伝子改変によるCAR-T細胞の最良化(4600字)
【第84回日本血液学会学術集会レポート】次世代型CAR-T細胞療法の開発動向――遺伝子改変によるCAR-T細胞の最良化(4600字)
腫瘍(オンコロジー)>造血器
慶應義塾大学医学部 先端医科学研究所 がん免疫研究部門 教授/講演当時: 愛知県がんセンター研究所 腫瘍免疫応答研究分野 分野長
籠谷 勇紀先生医師・歯科医師限定【第55回日本てんかん学会レポート】 国内初・大学病院てんかん科の挑戦―11年間の足跡(2000字)
【第55回日本てんかん学会レポート】 国内初・大学病院てんかん科の挑戦―11年間の足跡(2000字)
脳神経系疾患>その他
その他>検査
東北大学大学院医学系研究科 てんかん学分野 教授
中里 信和先生医師・歯科医師限定【第81回日本癌学会レポート】Non-MDの立場から見たがん研究の意義と課題(3600字)
【第81回日本癌学会レポート】Non-MDの立場から見たがん研究の意義と課題(3600字)
腫瘍(オンコロジー)>その他
公益財団法人がん研究会 がん化学療法センター 分子生物治療研究部 部長
清宮 啓之先生医師・歯科医師限定【第81回日本癌学会レポート】がん専門病院における新型コロナウイルス感染症への取り組み(3000字)
【第81回日本癌学会レポート】がん専門病院における新型コロナウイルス感染症への取り組み(3000字)
腫瘍(オンコロジー)>その他
感染症>ウイルス性
その他>ワクチン
国立研究開発法人 国立がん研究センター中央病院 感染症部/感染制御室 医師
岩田 敏先生医師・歯科医師限定【第81回日本癌学会レポート】がんサバイバーシップを支えるスマホアプリを用いた心理療法(3700字)
【第81回日本癌学会レポート】がんサバイバーシップを支えるスマホアプリを用いた心理療法(3700字)
その他>その他
国立がん研究センター がん対策研究所/国立がん研究センター中央病院 支持療法開発センター長
内富 庸介先生医師・歯科医師限定【第84回日本血液学会レポート】同種造血幹細胞移植に対するCOVD-19ワクチンの有効性――低抗体価リスク因子、中和抗体薬との併用は(3800字)
【第84回日本血液学会レポート】同種造血幹細胞移植に対するCOVD-19ワクチンの有効性――低抗体価リスク因子、中和抗体薬との併用は(3800字)
腫瘍(オンコロジー)>造血器
血液・リンパ疾患>血液
血液・リンパ疾患>リンパ
感染症>ウイルス性
その他>ワクチン
久留米大学医学部 内科学講座 血液・腫瘍内科部門 主任教授
長藤 宏司先生医師・歯科医師限定【第55回日本てんかん学会レポート】深層学習を用いたてんかん脳磁図の自動診断(2000字)
【第55回日本てんかん学会レポート】深層学習を用いたてんかん脳磁図の自動診断(2000字)
脳神経系疾患>その他
その他>検査
大阪大学大学院医学系研究科 脳機能診断再建学共同研究講座 特任教授
平田 雅之先生医師・歯科医師限定【第84回日本血液学会レポート】COVID-19パンデミックが血液疾患の診断数に与えた影響――持続的減少をきたしたITPと特発性再生不良性貧血(2300字)
【第84回日本血液学会レポート】COVID-19パンデミックが血液疾患の診断数に与えた影響――持続的減少をきたしたITPと特発性再生不良性貧血(2300字)
血液・リンパ疾患>血液
免疫系疾患>自己免疫疾患
感染症>ウイルス性
慶應義塾大学医学部 血液内科 専任講師
櫻井 政寿先生医師・歯科医師限定【第84回日本血液学会レポート】挙児希望のある慢性骨髄性白血病(CML)患者の治療(4100字)
【第84回日本血液学会レポート】挙児希望のある慢性骨髄性白血病(CML)患者の治療(4100字)
腫瘍(オンコロジー)>造血器
その他>周産期
医療法人 菊郷会 愛育病院 血液内科・血液病センター 血液病センター長
近藤 健先生医師・歯科医師限定【第81回日本癌学会レポート】遺族調査からみた進行がんとの共生――がん患者の人生最終段階における実態把握(3500字)
【第81回日本癌学会レポート】遺族調査からみた進行がんとの共生――がん患者の人生最終段階における実態把握(3500字)
腫瘍(オンコロジー)>その他
国立がん研究センター がん対策研究所 がん医療支援部 研究員
中澤 葉宇子先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会学術集会レポート】肝硬変患者における門脈肺高血圧症の診断――コホート研究によるスクリーニング要素の探求(2500字)
【第7回日本肺高血圧・肺循環学会学術集会レポート】肝硬変患者における門脈肺高血圧症の診断――コホート研究によるスクリーニング要素の探求(2500字)
消化器疾患>肝胆膵
心臓血管疾患>血管
日本医科大学付属病院 消化器内科 准教授
厚川 正則先生医師・歯科医師限定【第80回日本癌学会レポート】がんと生きる高齢者の介護予防――「高齢者機能評価(GA)」が必須の手法に(3500字)
【第80回日本癌学会レポート】がんと生きる高齢者の介護予防――「高齢者機能評価(GA)」が必須の手法に(3500字)
腫瘍(オンコロジー)>呼吸器
腫瘍(オンコロジー)>その他
その他>その他
東京都健康長寿医療センター 呼吸器内科部長
山本 寛先生医師・歯科医師限定【第55回日本てんかん学会学術集会レポート】大規模災害とコミュニティのレジリエンス――精神医学の視点から(2800字)
【第55回日本てんかん学会学術集会レポート】大規模災害とコミュニティのレジリエンス――精神医学の視点から(2800字)
精神疾患
脳神経系疾患>その他
感染症>ウイルス性
東北大学大学院医学系研究科 精神神経学分野 教授
富田 博秋先生医師・歯科医師限定【第81回日本癌学会レポート】HPVワクチン積極的勧奨再開・キャッチアップ接種で子宮頸がんリスクはどう変わるか――大阪大学のシミュレーション結果、今後の研究計画(4500字)
【第81回日本癌学会レポート】HPVワクチン積極的勧奨再開・キャッチアップ接種で子宮頸がんリスクはどう変わるか――大阪大学のシミュレーション結果、今後の研究計画(4500字)
腫瘍(オンコロジー)>泌尿生殖器
その他>ワクチン
大阪大学大学院医学系研究科 産科学婦人科学教室 講師
上田 豊先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会レポート】診断に苦慮し治療に至った領域横断的な肺高血圧症――単剤治療で様子を見るべき症例は(3400字)
【第7回日本肺高血圧・肺循環学会レポート】診断に苦慮し治療に至った領域横断的な肺高血圧症――単剤治療で様子を見るべき症例は(3400字)
心臓血管疾患>血管
免疫系疾患>自己免疫疾患
国際医療福祉大学 医学部 循環器内科学 教授/国際医療福祉大学 成田病院 循環器内科
杉村 宏一郎先生医師・歯科医師限定【第62回日本呼吸器学会レポート】デジタルデバイスを活用したオンライン診療――呼吸器内科診療はどう変わる?(3500字)
【第62回日本呼吸器学会レポート】デジタルデバイスを活用したオンライン診療――呼吸器内科診療はどう変わる?(3500字)
呼吸器疾患
その他>その他
社会医療法人春回会井上病院 院長
吉嶺 裕之先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会学術集会レポート】医療画像認識におけるAIの可能性(2800字)
【第7回日本肺高血圧・肺循環学会学術集会レポート】医療画像認識におけるAIの可能性(2800字)
心臓血管疾患>血管
その他>検査
徳島大学病院 循環器内科 講師
楠瀬 賢也先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会レポート】肺高血圧症治療の現状――PAH、CTEPHに対する診断・治療、肺移植の現状(4500字)
【第7回日本肺高血圧・肺循環学会レポート】肺高血圧症治療の現状――PAH、CTEPHに対する診断・治療、肺移植の現状(4500字)
心臓血管疾患>血管
心臓血管疾患>心臓
免疫系疾患>自己免疫疾患
東京大学医学部附属病院 高度心不全治療センター 准教授・センター長
波多野 将先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会レポート】医療用AIの現状と今後の可能性――診断や予後予測への応用(2600字)
【第7回日本肺高血圧・肺循環学会レポート】医療用AIの現状と今後の可能性――診断や予後予測への応用(2600字)
呼吸器疾患
その他>検査
その他>その他
名古屋大学医学部附属病院メディカルITセンター 特任助教/特定国立研究開発法人理化学研究所 画像情報処理研究チーム 客員研究員
古川 大記先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会学術集会レポート】時間経過に伴い複数の病態を合併するCTD-PH――多分野集学的検討の重要性を事例とともに解説(4200字)
【第7回日本肺高血圧・肺循環学会学術集会レポート】時間経過に伴い複数の病態を合併するCTD-PH――多分野集学的検討の重要性を事例とともに解説(4200字)
心臓血管疾患>心臓
免疫系疾患>自己免疫疾患
香川大学医学部・医学系研究科 血液・免疫・呼吸器内科学 助教
中島 崇作先生医師・歯科医師限定【第7回日本肺高血圧症・肺循環学会レポート】膠原病に伴う肺高血圧症診療の進歩と将来展望(4600字)
【第7回日本肺高血圧症・肺循環学会レポート】膠原病に伴う肺高血圧症診療の進歩と将来展望(4600字)
心臓血管疾患>心臓
免疫系疾患>自己免疫疾患
日本医科大学大学院医学研究科 アレルギー膠原病内科学分野 教授
桑名 正隆先生医師・歯科医師限定【第55回日本てんかん学会レポート】限局性皮質異形成II型に対するシロリムスの有効性・安全性に関する医師主導治験(FCDS-01)――第III相試験 2023年度に開始(2800字)
【第55回日本てんかん学会レポート】限局性皮質異形成II型に対するシロリムスの有効性・安全性に関する医師主導治験(FCDS-01)――第III相試験 2023年度に開始(2800字)
脳神経系疾患>その他
昭和大学医学部小児科学講座 教授/昭和大学病院 てんかん診療センター センター長
加藤 光広先生医師・歯科医師限定【第59回日本癌治療学会レポート】食道がんにおけるロボット支援手術の現状と展望(1800字)
【第59回日本癌治療学会レポート】食道がんにおけるロボット支援手術の現状と展望(1800字)
腫瘍(オンコロジー)>消化器
国立がん研究センター中央病院 食道外科長
大幸 宏幸先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】前立腺がんの予後予測因子――予後不良群への治療選択は?(3200字)
【第109回日本泌尿器科学会レポート】前立腺がんの予後予測因子――予後不良群への治療選択は?(3200字)
腫瘍(オンコロジー)>泌尿生殖器
名古屋大学大学院医学系研究科 泌尿器科学教室 准教授
加藤 真史先生医師・歯科医師限定【第65回日本糖尿病学会レポート】糖尿病治療の進歩がもたらしたもの――スティグマ克服に向けた発信を(3000字)
【第65回日本糖尿病学会レポート】糖尿病治療の進歩がもたらしたもの――スティグマ克服に向けた発信を(3000字)
内分泌系疾患>糖尿病
神戸大学大学院医学研究科 糖尿病・内分泌内科学部門 准教授
廣田 勇士先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会レポート】肺高血圧症における腸内細菌叢の異常――エンドトキシン、TMAOとの関連(3200字)
【第7回日本肺高血圧・肺循環学会レポート】肺高血圧症における腸内細菌叢の異常――エンドトキシン、TMAOとの関連(3200字)
心臓血管疾患>血管
医薬基盤・健康・栄養研究所 ワクチンマテリアルプロジェクト プロジェクト研究員
重城 喬行先生医師・歯科医師限定【第65回日本腎臓学会レポート】慢性腎臓病におけるSGLT2阻害薬の有効性――腎・心不全アウトカム試験のエビデンスを踏まえた検討(3200字)
【第65回日本腎臓学会レポート】慢性腎臓病におけるSGLT2阻害薬の有効性――腎・心不全アウトカム試験のエビデンスを踏まえた検討(3200字)
泌尿生殖器疾患>腎臓
順天堂大学医学部附属順天堂医院 腎・高血圧内科 先任准教授
合田 朋仁先生医師・歯科医師限定【第65回日本糖尿病学会レポート】K-ATPチャネルの構造解明――5年半の研究を振り返って(3900字)
【第65回日本糖尿病学会レポート】K-ATPチャネルの構造解明――5年半の研究を振り返って(3900字)
内分泌系疾患>糖尿病
その他>その他
京都大学 名誉教授/公益財団法人田附興風会 医学研究所北野病院 理事長
稲垣 暢也先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会レポート】肺高血圧症の新たな病態解明と治療薬の展望――肺血管リモデリングへの直接介入(3300字)
【第7回日本肺高血圧・肺循環学会レポート】肺高血圧症の新たな病態解明と治療薬の展望――肺血管リモデリングへの直接介入(3300字)
心臓血管疾患>血管
京都府立医科大学 長寿・地域疫学講座 教授
池田 宏二先生医師・歯科医師限定【第55回日本てんかん学会レポート】組織診断はLEAT概念のゴールドスタンダードとなりうるか?(3600字)
【第55回日本てんかん学会レポート】組織診断はLEAT概念のゴールドスタンダードとなりうるか?(3600字)
脳神経系疾患>その他
秋田県立循環器・脳脊髄センター 臨床病理部 部長、研究所 脳血管研究センター 脳神経病理学研究部 部長
宮田 元先生医師・歯科医師限定【第66回日本リウマチ学会レポート】変形性関節症の薬物療法におけるデュロキセチンとオピオイドの位置づけ――海外と日本の比較(3500字)
【第66回日本リウマチ学会レポート】変形性関節症の薬物療法におけるデュロキセチンとオピオイドの位置づけ――海外と日本の比較(3500字)
筋骨格系疾患>関節
その他>薬物治療
千葉大学医学部附属病院 整形外科 助教
瓦井 裕也先生医師・歯科医師限定【第66回日本リウマチ学会レポート】リウマチ性疾患患者におけるSARS-CoV-2ワクチン(2800字)
【第66回日本リウマチ学会レポート】リウマチ性疾患患者におけるSARS-CoV-2ワクチン(2800字)
免疫系疾患>自己免疫疾患
感染症>ウイルス性
その他>ワクチン
九州大学病院別府病院 内科 助教
木本 泰孝先生医師・歯科医師限定【第21回日本再生医療学会レポート】臨床応用段階に入った「心筋球移植」――数々の課題を克服した技術開発(2500字)
【第21回日本再生医療学会レポート】臨床応用段階に入った「心筋球移植」――数々の課題を克服した技術開発(2500字)
心臓血管疾患>心臓
その他>再生医療
慶應義塾大学医学部循環器内科 教授
福田 恵一先生医師・歯科医師限定【第66回日本リウマチ学会レポート】リウマチ膠原病における新たな分子標的治療――抗リウマチ薬の有効性と安全性(3300字)
【第66回日本リウマチ学会レポート】リウマチ膠原病における新たな分子標的治療――抗リウマチ薬の有効性と安全性(3300字)
免疫系疾患>自己免疫疾患
北里大学病院 膠原病・感染内科学 主任教授
山岡 邦宏先生医師・歯科医師限定【第94回日本胃癌学会レポート】胃がんに対する外科治療の課題と展望――薬物治療と組み合わせた個別化医療の発展(2400字)
【第94回日本胃癌学会レポート】胃がんに対する外科治療の課題と展望――薬物治療と組み合わせた個別化医療の発展(2400字)
腫瘍(オンコロジー)>消化器
がん研有明病院 病院長
佐野 武先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】mCRPCに対する薬剤の選択基準(3600字)
【第109回日本泌尿器科学会レポート】mCRPCに対する薬剤の選択基準(3600字)
腫瘍(オンコロジー)>泌尿生殖器
順天堂大学大学院医学研究科 泌尿器外科学 / 順天堂大学医学部附属順天堂医院 泌尿器科 准教授
永田 政義先生医師・歯科医師限定【第59回日本癌治療学会レポート】大腸手術におけるアウトカム向上を目指した医療機器の開発――AIによるナビゲーションシステムと酸素飽和度イメージング(2500字)
【第59回日本癌治療学会レポート】大腸手術におけるアウトカム向上を目指した医療機器の開発――AIによるナビゲーションシステムと酸素飽和度イメージング(2500字)
腫瘍(オンコロジー)>消化器
消化器疾患>消化管
国立がん研究センター東病院 医療機器開発推進部門 医療機器開発推進部 手術機器開発推進室 室長 / 国立がん研究センター東病院 大腸外科 医員
長谷川 寛先生医師・歯科医師限定【第65回日本腎臓学会レポート】分子標的治療薬・免疫チェックポイント阻害薬による腎障害の対策(2900字)
【第65回日本腎臓学会レポート】分子標的治療薬・免疫チェックポイント阻害薬による腎障害の対策(2900字)
腫瘍(オンコロジー)>その他
内分泌系疾患>その他
その他>薬物治療
名古屋大学医学部附属病院 化学療法部 部長(教授)
安藤 雄一先生医師・歯科医師限定【第119回日本内科学会レポート】腸内細菌beyond the gut――迷走神経反射を介した腸管の恒常性維持とその経路の解明(4400字)
【第119回日本内科学会レポート】腸内細菌beyond the gut――迷走神経反射を介した腸管の恒常性維持とその経路の解明(4400字)
免疫系疾患>自己免疫疾患
その他>その他
慶應義塾大学医学部 内科学(消化器)教室 教授
金井 隆典先生医師・歯科医師限定【第94回日本胃癌学会レポート】若手医師が臨床系英文論文を書く際のポイント――自身の経験をもとに考察(3200字)
【第94回日本胃癌学会レポート】若手医師が臨床系英文論文を書く際のポイント――自身の経験をもとに考察(3200字)
その他>その他
名古屋大学大学院医学系研究科 消化器外科学 講師
神田 光郎先生医師・歯科医師限定【第119回日本内科学会レポート】iPS細胞を用いたパーキンソン病治療(3800字)
【第119回日本内科学会レポート】iPS細胞を用いたパーキンソン病治療(3800字)
脳神経系疾患>その他
京都大学 iPS細胞研究所(CiRA) 所長
髙橋 淳先生医師・歯科医師限定【第94回日本胃癌学会レポート】胃がん患者に対する薬物療法の現状と今後の展望――他がん種の臨床成績を踏まえて(3000字)
【第94回日本胃癌学会レポート】胃がん患者に対する薬物療法の現状と今後の展望――他がん種の臨床成績を踏まえて(3000字)
腫瘍(オンコロジー)>消化器
東京大学医科学研究所附属病院 腫瘍・総合内科教授/診療科長
朴 成和先生医師・歯科医師限定【第119回日本内科学会レポート】膠原病における分子標的薬の可能性(2500字)
【第119回日本内科学会レポート】膠原病における分子標的薬の可能性(2500字)
免疫系疾患>自己免疫疾患
京都大学大学院医学研究科 内科学講座臨床免疫学 教授
森信 暁雄先生医師・歯科医師限定【第119回日本内科学会レポート】COVID-19による心臓・肺血管の後遺症「CV Long COVID」――心筋炎や肺塞栓症患者の特徴は(2000字)
【第119回日本内科学会レポート】COVID-19による心臓・肺血管の後遺症「CV Long COVID」――心筋炎や肺塞栓症患者の特徴は(2000字)
心臓血管疾患>血管
心臓血管疾患>心臓
感染症>ウイルス性
日本大学医学部内科学系循環器内科学分野 助教
村田 伸弘先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】腎移植後の前立腺がんや尿路上皮がんの発症とその治療(2300字)
【第109回日本泌尿器科学会レポート】腎移植後の前立腺がんや尿路上皮がんの発症とその治療(2300字)
腫瘍(オンコロジー)>泌尿生殖器
泌尿生殖器疾患>腎臓
岡山大学大学院医歯薬学総合研究科 泌尿器病態学 准教授
荒木 元朗先生医師・歯科医師限定【第59回日本癌治療学会学術集会レポート】胃がん周術期化学療法の現状と将来展望――各国における臨床試験の結果を踏まえて(3400字)
【第59回日本癌治療学会学術集会レポート】胃がん周術期化学療法の現状と将来展望――各国における臨床試験の結果を踏まえて(3400字)
腫瘍(オンコロジー)>消化器
がん研有明病院 消化器化学療法科副部長
高張 大亮先生医師・歯科医師限定【第53回日本動脈硬化学会レポート】がん関連血栓症の発症機序とバイオマーカーの使い分け(4500字)
【第53回日本動脈硬化学会レポート】がん関連血栓症の発症機序とバイオマーカーの使い分け(4500字)
腫瘍(オンコロジー)>その他
心臓血管疾患>血管
金沢大学大学院 医薬保健学総合研究科 保健学専攻・医療科学領域病態検査学講座 教授 / 日本医科大学付属病院 血液内科 客員教授
森下 英理子先生医師・歯科医師限定【第62回日本呼吸器学会レポート】COVID-19肺炎中等症IIの呼吸管理(人工呼吸に至るまで)(3600字)
【第62回日本呼吸器学会レポート】COVID-19肺炎中等症IIの呼吸管理(人工呼吸に至るまで)(3600字)
呼吸器疾患
感染症>ウイルス性
聖路加国際病院 呼吸器センター 管理医長
西村 直樹先生医師・歯科医師限定【第119回日本内科学会レポート】慢性腎臓病(CKD)対策における地域連携・多職種連携(3100字)
【第119回日本内科学会レポート】慢性腎臓病(CKD)対策における地域連携・多職種連携(3100字)
泌尿生殖器疾患>腎臓
島根大学医学部附属病院 腎臓内科 診療教授・診療科長/血液浄化治療部 部長
伊藤 孝史先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】m0CRPC に対する新規アンドロゲン受容体シグナル阻害薬(ARAT)3 剤の使用意義(2200字)
【第109回日本泌尿器科学会レポート】m0CRPC に対する新規アンドロゲン受容体シグナル阻害薬(ARAT)3 剤の使用意義(2200字)
腫瘍(オンコロジー)>泌尿生殖器
札幌医科大学医学部 泌尿器科講座 講師
橋本 浩平先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】オリゴ転移前立腺がんの診断と治療のトレンド――その定義と臨床的意義とは(2500字)
【第109回日本泌尿器科学会レポート】オリゴ転移前立腺がんの診断と治療のトレンド――その定義と臨床的意義とは(2500字)
腫瘍(オンコロジー)>泌尿生殖器
京都大学医学研究科泌尿器科学 教授
小林 恭先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】前立腺がんの手術療法と放射線療法の未来予想――超寡分割照射の可能性、“漏れない手術”に向けた技術向上(3100字)
【第109回日本泌尿器科学会レポート】前立腺がんの手術療法と放射線療法の未来予想――超寡分割照射の可能性、“漏れない手術”に向けた技術向上(3100字)
腫瘍(オンコロジー)>泌尿生殖器
関西医科大学 腎泌尿器外科学 教授
木下 秀文先生医師・歯科医師限定【第53回日本動脈硬化学会レポート】臨床研究から考える血糖コントロールと動脈硬化対策(4000字)
【第53回日本動脈硬化学会レポート】臨床研究から考える血糖コントロールと動脈硬化対策(4000字)
心臓血管疾患>血管
内分泌系疾患>糖尿病
千葉大学大学院医学研究院 内分泌代謝・血液・老年内科学 教授 / 千葉大学医学部附属病院 病院長 / 千葉大学 副学長
横手 幸太郎先生医師・歯科医師限定【第74回日本胸部外科学会レポート】局所進行胸部食道がんの集学的治療戦略――T3-T4ボーダーライン症例、T4症例に対する治療戦略と検証結果(2500字)
【第74回日本胸部外科学会レポート】局所進行胸部食道がんの集学的治療戦略――T3-T4ボーダーライン症例、T4症例に対する治療戦略と検証結果(2500字)
腫瘍(オンコロジー)>消化器
がん研有明病院 副院長/食道外科部長/消化器外科部長
渡邊 雅之先生医師・歯科医師限定【インタビュー】病棟カルテの書き方――悩める医学生・初期研修医へのメッセージ(若手指導医の視点から)(4500字)
【インタビュー】病棟カルテの書き方――悩める医学生・初期研修医へのメッセージ(若手指導医の視点から)(4500字)
その他>その他
岡山大学病院 総合内科・総合診療科/岡山大学学術研究院 医歯薬学域 県北西部総合診療医学講座 助教
大塚 勇輝先生医師・歯科医師限定【第19回日本臨床腫瘍学会レポート】多職種による就労支援の体制づくり――がん薬物療法専門医の視点から(2900字)
【第19回日本臨床腫瘍学会レポート】多職種による就労支援の体制づくり――がん薬物療法専門医の視点から(2900字)
腫瘍(オンコロジー)>呼吸器
腫瘍(オンコロジー)>その他
神奈川県立循環器呼吸器病センター 呼吸器内科 臨床研究室 医長
池田 慧先生医師・歯科医師限定【第59回日本癌治療学会レポート】RET融合遺伝子陽性肺がんに対する治療開発の現状と課題――臨床試験の症例を踏まえて(2300字)
【第59回日本癌治療学会レポート】RET融合遺伝子陽性肺がんに対する治療開発の現状と課題――臨床試験の症例を踏まえて(2300字)
腫瘍(オンコロジー)>呼吸器
鳥取大学医学部附属病院 呼吸器内科・膠原病内科 特任助教
阪本 智宏先生医師・歯科医師限定【第59回日本癌治療学会レポート】泌尿器科ロボット支援手術の現状と未来――次世代教育の課題(2700字)
【第59回日本癌治療学会レポート】泌尿器科ロボット支援手術の現状と未来――次世代教育の課題(2700字)
腫瘍(オンコロジー)>泌尿生殖器
国立がん研究センター東病院 泌尿器・後腹膜腫瘍科 科長
増田 均先生医師・歯科医師限定【第59回日本癌治療学会レポート】進行非小細胞肺がんの分子標的治療の耐性と克服戦略(4500字)
【第59回日本癌治療学会レポート】進行非小細胞肺がんの分子標的治療の耐性と克服戦略(4500字)
腫瘍(オンコロジー)>呼吸器
国立がん研究センター 東病院 呼吸器内科 医長
葉 清隆先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】nmCRPCの治療――画像診断の必要性、新規AR薬投与の意義は?(2400字)
【第109回日本泌尿器科学会レポート】nmCRPCの治療――画像診断の必要性、新規AR薬投与の意義は?(2400字)
腫瘍(オンコロジー)>泌尿生殖器
東京慈恵会医科大学 泌尿器科 診療部長/教授
木村 高弘先生医師・歯科医師限定【第109回日本泌尿器科学会レポート】オリゴ転移前立腺がんに対する治療――臨床試験データから考える現状とこれから(3300字)
【第109回日本泌尿器科学会レポート】オリゴ転移前立腺がんに対する治療――臨床試験データから考える現状とこれから(3300字)
腫瘍(オンコロジー)>泌尿生殖器
藤沢市民病院 泌尿器科 専門医長/横浜市立大学 客員教授/横浜市立大学附属市民総合医療センター泌尿器・腎移植科 非常勤診療医
三好 康秀先生医師・歯科医師限定【第19回日本臨床腫瘍学会レポート】COVID-19が肺がん診療に及ぼした影響――日本肺癌学会、日本対がん協会の調査報告をもとに(2000字)
【第19回日本臨床腫瘍学会レポート】COVID-19が肺がん診療に及ぼした影響――日本肺癌学会、日本対がん協会の調査報告をもとに(2000字)
腫瘍(オンコロジー)>呼吸器
感染症>ウイルス性
大分大学医学部 呼吸器・乳腺外科学講座 教授
杉尾 賢二先生医師・歯科医師限定【第59回日本癌治療学会レポート】切除不能胃がんに対する薬物療法の新たな治療戦略(3900字)
【第59回日本癌治療学会レポート】切除不能胃がんに対する薬物療法の新たな治療戦略(3900字)
腫瘍(オンコロジー)>消化器
国立がん研究センター東病院 消化管内科
川添 彬人先生医師・歯科医師限定【第53回日本動脈硬化学会レポート】GLP-1、GIP、DPP-4阻害薬の抗動脈硬化作用(3700字)
【第53回日本動脈硬化学会レポート】GLP-1、GIP、DPP-4阻害薬の抗動脈硬化作用(3700字)
心臓血管疾患>血管
その他>薬物治療
社会医療法人ジャパンメディカルアライアンス 海老名総合病院 糖尿病センター センター長
平野 勉先生医師・歯科医師限定【第62回日本肺癌学会レポート】血液マイクロRNAを用いた次世代型がん検診(3000字)
【第62回日本肺癌学会レポート】血液マイクロRNAを用いた次世代型がん検診(3000字)
腫瘍(オンコロジー)>その他
東京医科大学医学総合研究所 分子細胞治療研究部門 教授
落谷 孝広先生医師・歯科医師限定【第59回日本癌治療学会レポート】がんの近赤外光線免疫療法(NIR-PIT)――メカニズムや課題、今後の展望とは(3000字)
【第59回日本癌治療学会レポート】がんの近赤外光線免疫療法(NIR-PIT)――メカニズムや課題、今後の展望とは(3000字)
腫瘍(オンコロジー)>その他
アメリカ国立衛生研究所(NIH)/アメリカ国立がん研究所(NCI)主任研究員
小林 久隆先生医師・歯科医師限定【第7回日本肺高血圧・肺循環学会レポート】膠原病性肺動脈性肺高血圧症の病態形成をIL-6シグナルから考える(3400字)
【第7回日本肺高血圧・肺循環学会レポート】膠原病性肺動脈性肺高血圧症の病態形成をIL-6シグナルから考える(3400字)
呼吸器疾患
心臓血管疾患>心臓
免疫系疾患>自己免疫疾患
国立循環器病研究センター研究所 血管生理学部 部長
中岡 良和先生医師・歯科医師限定【第65回日本糖尿病学会レポート】糖尿病を持って生活する人々とスティグマ――発生の構造的要因と改善を目指したアドボカシー(3300字)
【第65回日本糖尿病学会レポート】糖尿病を持って生活する人々とスティグマ――発生の構造的要因と改善を目指したアドボカシー(3300字)
内分泌系疾患>糖尿病
東京大学大学院医学系研究科 公共健康医学専攻 行動社会医学講座 教授
橋本 英樹先生医師・歯科医師限定【第62回日本肺癌学会レポート】進行非小細胞肺がんにおける免疫療法――各種薬剤が免疫系にもたらす影響と将来的な可能性(3300字)
【第62回日本肺癌学会レポート】進行非小細胞肺がんにおける免疫療法――各種薬剤が免疫系にもたらす影響と将来的な可能性(3300字)
腫瘍(オンコロジー)>呼吸器
独立行政法人 国立病院機構 四国がんセンター 呼吸器内科/臨床研究センター長
上月 稔幸先生医師・歯科医師限定【第53回日本動脈硬化学会レポート】重症COVID-19における血栓症と抗凝固療法――ROTEMの有用性に関する検討(4300字)
【第53回日本動脈硬化学会レポート】重症COVID-19における血栓症と抗凝固療法――ROTEMの有用性に関する検討(4300字)
感染症>ウイルス性
名古屋大学医学部附属病院 循環器内科 病院助教
平岩 宏章先生医師・歯科医師限定【第51回日本皮膚免疫アレルギー学会レポート】果物や野菜によるアナフィラキシーのメカニズムと見分け方(4000字)
【第51回日本皮膚免疫アレルギー学会レポート】果物や野菜によるアナフィラキシーのメカニズムと見分け方(4000字)
免疫系疾患>アレルギー
昭和大学医学部皮膚科学講座 主任教授/昭和大学病院皮膚科 診療科長
猪又 直子先生医師・歯科医師限定【第51回日本皮膚免疫アレルギー学会レポート】アドレナリン性蕁麻疹の診断と治療――鑑別に難渋した症例を含めて(4500字)
【第51回日本皮膚免疫アレルギー学会レポート】アドレナリン性蕁麻疹の診断と治療――鑑別に難渋した症例を含めて(4500字)
免疫系疾患>アレルギー
大阪医科薬科大学 皮膚科 准教授/同大学病院 アレルギーセンター 副センター長
福永 淳先生医師・歯科医師限定【第51回日本皮膚免疫アレルギー学会レポート】食品・薬剤添加物アナフィラキシー 見逃さないポイントを事例とともに紹介(5000字)
【第51回日本皮膚免疫アレルギー学会レポート】食品・薬剤添加物アナフィラキシー 見逃さないポイントを事例とともに紹介(5000字)
免疫系疾患>アレルギー
神戸市立西神戸医療センター 皮膚科 部長代行
鷲尾 健先生医師・歯科医師限定【第80回日本癌学会レポート】リキッドバイオプシーを用いたがんゲノム医療の新展開(4900字)
【第80回日本癌学会レポート】リキッドバイオプシーを用いたがんゲノム医療の新展開(4900字)
腫瘍(オンコロジー)>その他
国立がん研究センター東病院 消化管内科 科長
吉野 孝之先生医師・歯科医師限定【インタビュー】持続グルコースモニタリングで糖尿病患者のQOL向上――非侵襲デバイス実用化にはさらなるブレイクスルー必要(1250字)
【インタビュー】持続グルコースモニタリングで糖尿病患者のQOL向上――非侵襲デバイス実用化にはさらなるブレイクスルー必要(1250字)
内分泌系疾患>糖尿病
国立国際医療研究センター研究所 糖尿病研究センター長/日本糖尿病学会理事長
植木 浩二郎先生医師・歯科医師限定【インタビュー】高齢糖尿病患者の食事療法、虚弱・筋肉量減少予防優先を――難しいエビデンスづくり(1200字)
【インタビュー】高齢糖尿病患者の食事療法、虚弱・筋肉量減少予防優先を――難しいエビデンスづくり(1200字)
筋骨格系疾患>筋
泌尿生殖器疾患>腎臓
内分泌系疾患>糖尿病
国立国際医療研究センター研究所 糖尿病研究センター長/日本糖尿病学会理事長
植木 浩二郎先生医師・歯科医師限定【インタビュー】行動変容に必要なデジタル化、日本の議論に欠けている「データは患者のもの」の視点(1000字)
【インタビュー】行動変容に必要なデジタル化、日本の議論に欠けている「データは患者のもの」の視点(1000字)
内分泌系疾患>糖尿病
国立国際医療研究センター研究所 糖尿病研究センター長/日本糖尿病学会理事長
植木 浩二郎先生医師・歯科医師限定【第53回日本動脈硬化学会レポート】COVID-19と生活習慣病・心血管不全の関連――重症化リスク、ワクチン接種との関係は(3400字)
【第53回日本動脈硬化学会レポート】COVID-19と生活習慣病・心血管不全の関連――重症化リスク、ワクチン接種との関係は(3400字)
心臓血管疾患>血管
心臓血管疾患>心臓
感染症>ウイルス性
佐賀大学医学部循環器内科教授・内科主任教授
野出 孝一先生医師・歯科医師限定【第119回日本内科学会レポート】高齢者糖尿病の診療ポイント――認知機能障害やフレイルとの関連、対策、薬物治療について(4200字)
【第119回日本内科学会レポート】高齢者糖尿病の診療ポイント――認知機能障害やフレイルとの関連、対策、薬物治療について(4200字)
筋骨格系疾患>筋
内分泌系疾患>糖尿病
東京都健康長寿医療センター 副院長/内科総括部長/フレイル予防センター長
荒木 厚先生医師・歯科医師限定【セミナーレポート】「ハートノート」がつなぐ心臓病診療――心不全による再入院を減らすための課題、多様化が必要な治療の目標/場所/従事者(3700字)
【セミナーレポート】「ハートノート」がつなぐ心臓病診療――心不全による再入院を減らすための課題、多様化が必要な治療の目標/場所/従事者(3700字)
心臓血管疾患>心臓
大阪大学 大学院医学系研究科 循環器内科学 教授
坂田 泰史先生医師・歯科医師限定【セミナーレポート】患者参加の電子診療記録の可能性――現状の課題、プラットフォームを介した医療情報共有システムのメリット(4700字)
【セミナーレポート】患者参加の電子診療記録の可能性――現状の課題、プラットフォームを介した医療情報共有システムのメリット(4700字)
心臓血管疾患>心臓
国立病院機構大阪医療センター 院長 / 一般社団法人健康医療クロスイノベーションラボ 理事
松村 泰志先生医師・歯科医師限定【第119回日本内科学会レポート】膠原病における間質性肺疾患(ILD)診断・治療のポイント――全身性強皮症や多発性筋炎/皮膚筋炎での特徴とは(6800字)
【第119回日本内科学会レポート】膠原病における間質性肺疾患(ILD)診断・治療のポイント――全身性強皮症や多発性筋炎/皮膚筋炎での特徴とは(6800字)
呼吸器疾患
免疫系疾患>自己免疫疾患
日本医科大学大学院医学研究科 アレルギー膠原病内科学分野 大学院教授 / 日本医科大学付属病院 リウマチ・膠原病内科 部長 / 強皮症・筋炎先進医療センター センター長
桑名 正隆先生医師・歯科医師限定【第19回日本臨床腫瘍学会レポート】COVID-19流行のがん検診・がん診療への影響(1800字)
【第19回日本臨床腫瘍学会レポート】COVID-19流行のがん検診・がん診療への影響(1800字)
腫瘍(オンコロジー)>その他
感染症>ウイルス性
国立がん研究センターがん対策研究所 検診研究部検診実施管理研究室長/がん医療支援部検診実施管理支援室長
高橋 宏和先生医師・歯科医師限定【第80回日本癌学会レポート】COVID-19 流行下におけるがん外科診療――アンケート結果に基づく手術状況の変化(2900字)
【第80回日本癌学会レポート】COVID-19 流行下におけるがん外科診療――アンケート結果に基づく手術状況の変化(2900字)
腫瘍(オンコロジー)>その他
感染症>ウイルス性
群馬大学大学院医学系研究科 総合外科学講座 肝胆膵外科学分野 教授 / 群馬大学医学部附属病院 外科診療センター長
調 憲先生医師・歯科医師限定【第80回日本癌学会レポート】COVID-19が血液腫瘍患者に与える影響(2800字)
【第80回日本癌学会レポート】COVID-19が血液腫瘍患者に与える影響(2800字)
呼吸器疾患
腫瘍(オンコロジー)>その他
感染症>ウイルス性
神戸大学医学部附属病院 腫瘍・血液内科
小山 泰司先生医師・歯科医師限定【第74回日本胸部外科学会レポート】食道癌の低侵襲手術――小開胸で行うポイントは(3500字)
【第74回日本胸部外科学会レポート】食道癌の低侵襲手術――小開胸で行うポイントは(3500字)
千葉大学大学院医学研究院 先端応用外科学 教授 / 千葉大学医学部附属病院 食道・胃腸外科 科長
松原 久裕先生医師・歯科医師限定【プレスリリース紹介】糖尿病網膜症・黄斑浮腫の低侵襲早期診断法の確立とフェノフィブラートナノ粒子点眼による新規低侵襲治療法確立への可能性(2800字)
【プレスリリース紹介】糖尿病網膜症・黄斑浮腫の低侵襲早期診断法の確立とフェノフィブラートナノ粒子点眼による新規低侵襲治療法確立への可能性(2800字)
MedicalNoteExpert編集部医師・歯科医師限定【第80回日本癌学会レポート】EGFR遺伝子変異陽性肺がんにおける免疫応答の解明――EGFR遺伝子変異陽性で発現が変化するケモカイン(3300字)
【第80回日本癌学会レポート】EGFR遺伝子変異陽性肺がんにおける免疫応答の解明――EGFR遺伝子変異陽性で発現が変化するケモカイン(3300字)
厚生労働省大臣官房厚生科学課・国立がん研究センター東病院 呼吸器内科所属
杉山 栄里先生医師・歯科医師限定【第80回日本癌学会レポート】EGFR阻害剤に対する耐性機序の研究――ノーベル賞受賞のゲノム編集「CRISPR-Cas9」を使った予想外の発見とは(3500字)
【第80回日本癌学会レポート】EGFR阻害剤に対する耐性機序の研究――ノーベル賞受賞のゲノム編集「CRISPR-Cas9」を使った予想外の発見とは(3500字)
国立がん研究センター研究所 分子病理分野
小林 祥久先生医師・歯科医師限定【論文紹介】Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial
【論文紹介】Casirivimab and imdevimab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial
ダナ・ファーバー癌研究所 メディカルオンコロジー分野
郭 悠先生医師・歯科医師限定【第80回日本癌学会レポート】胃がんの転移に対するHER2標的アルファ線治療――腹膜播種モデル・肝転移モデルマウスを用いた治療実験(2700字)
【第80回日本癌学会レポート】胃がんの転移に対するHER2標的アルファ線治療――腹膜播種モデル・肝転移モデルマウスを用いた治療実験(2700字)
量子科学技術研究開発機構 重粒子線治療研究部 放射線がん生物学研究グループ 主任研究員
李 惠子先生医師・歯科医師限定【第59回日本癌治療学会レポート】腫瘍循環器学の現状と課題――がん治療関連心筋障害、心不全と血栓症を中心に(4400字)
【第59回日本癌治療学会レポート】腫瘍循環器学の現状と課題――がん治療関連心筋障害、心不全と血栓症を中心に(4400字)
国立がん研究センター中央病院 総合内科・循環器内科医長
岩佐 健史先生医師・歯科医師限定【ニュース】COVID-19レジストリ登録データを見える化――COVIREGI-JPダッシュボードを公開 国立国際医療研究センター
【ニュース】COVID-19レジストリ登録データを見える化――COVIREGI-JPダッシュボードを公開 国立国際医療研究センター
MedicalNoteExpert編集部医師・歯科医師限定【第80回日本癌学会レポート】消化管がんに対する新しい内視鏡治療の開発――光免疫療法や腫瘍溶解性ウイルス療法の可能性(3700字)
【第80回日本癌学会レポート】消化管がんに対する新しい内視鏡治療の開発――光免疫療法や腫瘍溶解性ウイルス療法の可能性(3700字)
国立がん研究センター東病院 消化管内視鏡科長・内視鏡センター長
矢野 友規先生医師・歯科医師限定【論文紹介】mRNA-1273 vaccine-induced antibodies maintain Fc effector functions across SARS-CoV-2 variants of concern
【論文紹介】mRNA-1273 vaccine-induced antibodies maintain Fc effector functions across SARS-CoV-2 variants of concern
ダナ・ファーバー癌研究所 メディカルオンコロジー分野
郭 悠先生医師・歯科医師限定【第43回日本高血圧学会レポート】新しい医療・学術領域としてのOnco-Hypertension――がん併存疾患としての高血圧を考える
【第43回日本高血圧学会レポート】新しい医療・学術領域としてのOnco-Hypertension――がん併存疾患としての高血圧を考える
香川大学医学部 薬理学教室 教授
西山 成先生医師・歯科医師限定【論文紹介】『3階以上の階段利用で心房細動リスクが低下――国循 吹田研究で明らかに』
【論文紹介】『3階以上の階段利用で心房細動リスクが低下――国循 吹田研究で明らかに』
MedicalNoteExpert編集部医師・歯科医師限定【第83回日本血液学会レポート】造血器腫瘍のプレシジョン医療 ――ゲノム情報の活用について(4500文字)
【第83回日本血液学会レポート】造血器腫瘍のプレシジョン医療 ――ゲノム情報の活用について(4500文字)
九州大学大学院医学研究院 プレシジョン医療学分野 教授
前田 高宏先生医師・歯科医師限定内分泌系疾患 関連ガイドライン一覧
内分泌系疾患 関連ガイドライン一覧
MedicalNoteExpert編集部医師・歯科医師限定【第80回日本癌学会レポート】食道がんにおける免疫チェックポイント阻害剤の臨床――適用承認の元となった臨床試験の結果と今後の展望(4700字)
【第80回日本癌学会レポート】食道がんにおける免疫チェックポイント阻害剤の臨床――適用承認の元となった臨床試験の結果と今後の展望(4700字)
国立がん研究センター中央病院 頭頸部・食道内科科長(消化管内科科長併任)
加藤 健先生医師・歯科医師限定【インタビュー】肺高血圧症を伴う全身性強皮症、増えつつある薬の選択肢――基礎疾患治療の進歩により肺高血圧症の発症抑制にも期待(800文字)
【インタビュー】肺高血圧症を伴う全身性強皮症、増えつつある薬の選択肢――基礎疾患治療の進歩により肺高血圧症の発症抑制にも期待(800文字)
日本医科大学 大学院医学研究科アレルギー膠原病内科学分野 大学院教授/日本医科大学付属病院 リウマチ・膠原病内科 部長/強皮症・筋炎先進医療センター センター長
桑名 正隆先生医師・歯科医師限定【インタビュー】肺高血圧症治療の展望――肺血管リモデリングを改善する新薬登場の可能性(700文字)
【インタビュー】肺高血圧症治療の展望――肺血管リモデリングを改善する新薬登場の可能性(700文字)
日本医科大学 大学院医学研究科アレルギー膠原病内科学分野 大学院教授/日本医科大学付属病院 リウマチ・膠原病内科 部長/強皮症・筋炎先進医療センター センター長
桑名 正隆先生医師・歯科医師限定【インタビュー】幅広い診療科の医師に伝えたい肺高血圧症発見のポイントとレジストリの重要性――「労作時の息切れから心エコー」の発想を(1900文字)
【インタビュー】幅広い診療科の医師に伝えたい肺高血圧症発見のポイントとレジストリの重要性――「労作時の息切れから心エコー」の発想を(1900文字)
日本医科大学 大学院医学研究科アレルギー膠原病内科学分野 大学院教授/日本医科大学付属病院 リウマチ・膠原病内科 部長/強皮症・筋炎先進医療センター センター長
桑名 正隆先生医師・歯科医師限定【インタビュー】肺高血圧症は「肺血管拡張薬」で治療成績が劇的に向上――高まる個別化医療の重要性、早期診断に向けた国際的な定義見直しも(1600文字)
【インタビュー】肺高血圧症は「肺血管拡張薬」で治療成績が劇的に向上――高まる個別化医療の重要性、早期診断に向けた国際的な定義見直しも(1600文字)
日本医科大学 大学院医学研究科アレルギー膠原病内科学分野 大学院教授/日本医科大学付属病院 リウマチ・膠原病内科 部長/強皮症・筋炎先進医療センター センター長
桑名 正隆先生医師・歯科医師限定【第70回日本アレルギー学会レポート】鼻アレルギー診療ガイドラインはどう変わったか――第9版の改訂ポイントを解説(4400字)
【第70回日本アレルギー学会レポート】鼻アレルギー診療ガイドラインはどう変わったか――第9版の改訂ポイントを解説(4400字)
国際医療福祉大学 大学院医学研究科 耳鼻咽喉科学 教授
岡野 光博先生医師・歯科医師限定【第43回日本高血圧学会レポート】慢性腎臓病と高血圧――腎臓内シグナル伝達系から病態を理解する(4400文字)
【第43回日本高血圧学会レポート】慢性腎臓病と高血圧――腎臓内シグナル伝達系から病態を理解する(4400文字)
帝京大学医学部内科学講座 腎臓研究室 教授
柴田 茂先生医師・歯科医師限定【第64回日本糖尿病学会レポート】糖尿病患者における大血管症のリスクファクター管理と予防(6500文字)
【第64回日本糖尿病学会レポート】糖尿病患者における大血管症のリスクファクター管理と予防(6500文字)
大阪大学大学院医学系研究科内分泌・代謝内科 講師
片上 直人先生医師・歯科医師限定【第70回日本アレルギー学会レポート】「春季以外」のアレルギー性鼻炎の疫学・治療――イネ、ブタクサなどの草本花粉の対策は植生地域・飛散特性の把握から(4700字)
【第70回日本アレルギー学会レポート】「春季以外」のアレルギー性鼻炎の疫学・治療――イネ、ブタクサなどの草本花粉の対策は植生地域・飛散特性の把握から(4700字)
千葉大学大学院医学研究院 耳鼻咽喉科・頭頸部腫瘍学 准教授/千葉大学医学部附属病院 耳鼻咽喉・頭頸部外科
米倉 修二先生医師・歯科医師限定【第70回日本アレルギー学会レポート】花粉やダニによるアレルギー性鼻炎の免疫療法、その効果や安全性――喘息や新規感作の抑制効果はあるのか(5200字)
【第70回日本アレルギー学会レポート】花粉やダニによるアレルギー性鼻炎の免疫療法、その効果や安全性――喘息や新規感作の抑制効果はあるのか(5200字)
山梨大学大学院総合研究部医学域 耳鼻咽喉科・頭頸部外科学 教授
櫻井 大樹先生医師・歯科医師限定【第43回日本高血圧学会レポート】withコロナ時代でのRA系阻害薬内服の意義、循環老廃物が引き起こすAT1活性化機構に対するAT1阻害薬の可能性(3800字)
【第43回日本高血圧学会レポート】withコロナ時代でのRA系阻害薬内服の意義、循環老廃物が引き起こすAT1活性化機構に対するAT1阻害薬の可能性(3800字)
大阪大学大学院医学系研究科 老年・総合内科学 准教授
山本 浩一先生医師・歯科医師限定【第43回日本高血圧学会レポート】高齢者高血圧へのCOVID-19の影響――ACE2欠損による老化、フレイルによるCOVID-19重症化(3000字)
【第43回日本高血圧学会レポート】高齢者高血圧へのCOVID-19の影響――ACE2欠損による老化、フレイルによるCOVID-19重症化(3000字)
大阪大学大学院医学系研究科 老年・総合内科学 准教授
山本 浩一先生医師・歯科医師限定【第59回日本癌治療学会レポート】子宮頸がん予防におけるHPVワクチン有効性の実証――日本人若年女性対象の大規模疫学研究の結果、積極的勧奨中止の影響とは(4000文字)
【第59回日本癌治療学会レポート】子宮頸がん予防におけるHPVワクチン有効性の実証――日本人若年女性対象の大規模疫学研究の結果、積極的勧奨中止の影響とは(4000文字)
新潟大学医学部産科婦人科学教室 准教授
関根 正幸先生医師・歯科医師限定【第59回日本癌治療学会レポート】母体の子宮頸がんの移行による小児がん――羊水を吸い込み肺がんを発症した2症例(2600字)
【第59回日本癌治療学会レポート】母体の子宮頸がんの移行による小児がん――羊水を吸い込み肺がんを発症した2症例(2600字)
国立がん研究センター中央病院 小児腫瘍科医長
荒川 歩先生医師・歯科医師限定【第59回日本癌治療学会レポート】粘膜免疫を介した、子宮頸がん治療ワクチンの開発(2300字)
【第59回日本癌治療学会レポート】粘膜免疫を介した、子宮頸がん治療ワクチンの開発(2300字)
日本大学 医学部産婦人科学系産婦人科学分野 主任教授
川名 敬先生医師・歯科医師限定【第59回日本癌治療学会レポート】中咽頭がんにおけるHPV感染(4000字)
【第59回日本癌治療学会レポート】中咽頭がんにおけるHPV感染(4000字)
東京大学医学部附属病院 耳鼻咽喉科・頭頸部外科
齊藤 祐毅先生医師・歯科医師限定【第59回日本癌治療学会レポート】国内外におけるHPVワクチンの現状――9価ワクチンの導入、今後の課題とは(3700字)
【第59回日本癌治療学会レポート】国内外におけるHPVワクチンの現状――9価ワクチンの導入、今後の課題とは(3700字)
国立研究開発法人 国立がん研究センター中央病院 感染症部長/感染制御室長
岩田 敏先生医師・歯科医師限定【第43回日本高血圧学会レポート】レニン・アンジオテンシン系阻害薬とCOVID-19罹患・重症化リスクとの関連(4300字)
【第43回日本高血圧学会レポート】レニン・アンジオテンシン系阻害薬とCOVID-19罹患・重症化リスクとの関連(4300字)
横浜市立大学医学部医学科 循環器・腎臓・高血圧内科学教室 主任教授/横浜市立大学附属病院 副病院長
田村 功一先生医師・歯科医師限定【インタビュー】社会的問題の「不育症」、抗リン脂質抗体症候群が原因なら治療可能――概念浸透がまず課題(800字)
【インタビュー】社会的問題の「不育症」、抗リン脂質抗体症候群が原因なら治療可能――概念浸透がまず課題(800字)
北海道大学大学院医学研究院 免疫・代謝内科学教室 教授
渥美 達也先生医師・歯科医師限定【インタビュー】生物学的製剤ベリムマブ、ループス腎炎の寛解導入療法に効果――有用性評価の可能性も(700字)
【インタビュー】生物学的製剤ベリムマブ、ループス腎炎の寛解導入療法に効果――有用性評価の可能性も(700字)
北海道大学大学院医学研究院 免疫・代謝内科学教室 教授
渥美 達也先生医師・歯科医師限定【インタビュー】全身性エリテマトーデス治療にも分子標的薬――アニフロルマブ米で承認も効果は一部患者に限定(400字)
【インタビュー】全身性エリテマトーデス治療にも分子標的薬――アニフロルマブ米で承認も効果は一部患者に限定(400字)
北海道大学大学院医学研究院 免疫・代謝内科学教室 教授
渥美 達也先生医師・歯科医師限定【インタビュー】関節リウマチ治療薬は生物学的製剤からJAK阻害薬へ――選択肢増、経口摂取がアドバンテージに(1000字)
【インタビュー】関節リウマチ治療薬は生物学的製剤からJAK阻害薬へ――選択肢増、経口摂取がアドバンテージに(1000字)
北海道大学大学院医学研究院 免疫・代謝内科学教室 教授
渥美 達也先生医師・歯科医師限定【学会レポート】限局性強皮症の診断――種類の鑑別、活動性の評価(5900字)
【学会レポート】限局性強皮症の診断――種類の鑑別、活動性の評価(5900字)
東京大学 皮膚科学教室 医局長
山下 尚志先生医師・歯科医師限定【学会レポート】CKDの病態を可視化するFunctional MRI――間質線維化や低酸素状態の評価が可能に(4100文字)
【学会レポート】CKDの病態を可視化するFunctional MRI――間質線維化や低酸素状態の評価が可能に(4100文字)
埼玉医科大学 医学部腎臓内科 教授
岡田 浩一先生医師・歯科医師限定【学会レポート】COVID-19の疫学的特徴――SARSとの違い、感染伝播の異質性、第5波収束の理由(4600文字)
【学会レポート】COVID-19の疫学的特徴――SARSとの違い、感染伝播の異質性、第5波収束の理由(4600文字)
東北大学大学院医学系研究科 微生物学分野 教授
押谷 仁先生医師・歯科医師限定【学会レポート】CPER法による簡便・迅速な新型コロナウイルス人工合成技術の確立――変異株にどう対応していくか(3300字)
【学会レポート】CPER法による簡便・迅速な新型コロナウイルス人工合成技術の確立――変異株にどう対応していくか(3300字)
北海道大学大学院医学研究院 病理系部門 微生物学免疫学分野 教授
福原 崇介先生医師・歯科医師限定【学会レポート】新型コロナウイルスの変異と免疫――時間とともに変化する中和抗体の質、新たな抗体「NT-193」への期待(3000字)
【学会レポート】新型コロナウイルスの変異と免疫――時間とともに変化する中和抗体の質、新たな抗体「NT-193」への期待(3000字)
国立感染症研究所 治療薬・ワクチン開発研究センター センター長
高橋 宜聖先生医師・歯科医師限定【学会レポート】COVID-19後遺症と治療のトピックス――治療薬の選択とそのエビデンス(4800字)
【学会レポート】COVID-19後遺症と治療のトピックス――治療薬の選択とそのエビデンス(4800字)
大阪大学大学院医学系研究科 感染制御医学講座 教授
忽那 賢志先生医師・歯科医師限定【学会レポート】SARS-CoV-2の感染メカニズム――フーリンとTMPRSS2による開裂活性化、変異株での特徴(2600字)
【学会レポート】SARS-CoV-2の感染メカニズム――フーリンとTMPRSS2による開裂活性化、変異株での特徴(2600字)
国立感染症研究所 ウイルス第三部 部長
竹田 誠先生医師・歯科医師限定【学会レポート】肝中心静脈閉塞症(SOS/VOD)の診断・治療――デフィブロタイド投与の症例紹介も含めて(2900字)
【学会レポート】肝中心静脈閉塞症(SOS/VOD)の診断・治療――デフィブロタイド投与の症例紹介も含めて(2900字)
福島県立医科大学 小児腫瘍内科 特任教授
菊田 敦先生医師・歯科医師限定【インタビュー】進歩する肺がんに対する薬物療法・放射線治療とのコンビネーション、サルベージ手術で拡大する根治切除の可能性――今後のエビデンス構築に期待(400字)
【インタビュー】進歩する肺がんに対する薬物療法・放射線治療とのコンビネーション、サルベージ手術で拡大する根治切除の可能性――今後のエビデンス構築に期待(400字)
広島大学病院 呼吸器外科 科長/教授
岡田 守人先生医師・歯科医師限定【学会レポート】新しい抗HER2薬の開発――ツカチニブやT-DXdの可能性(3000字)
【学会レポート】新しい抗HER2薬の開発――ツカチニブやT-DXdの可能性(3000字)
がん研有明病院 院長補佐/乳腺内科 部長
高野 利実先生医師・歯科医師限定【インタビュー】小型非小細胞肺がん手術 優越性試験でも区域切除が肺葉切除を上回る――2022年4月The Lancetに掲載されたJCOG0802研究とその経緯(1400字)
【インタビュー】小型非小細胞肺がん手術 優越性試験でも区域切除が肺葉切除を上回る――2022年4月The Lancetに掲載されたJCOG0802研究とその経緯(1400字)
広島大学病院 呼吸器外科 科長/教授
岡田 守人先生医師・歯科医師限定COVID-19 パンデミック禍における造血器腫瘍の治療――永寿総合病院のデータ・事例をもとに
COVID-19 パンデミック禍における造血器腫瘍の治療――永寿総合病院のデータ・事例をもとに
永寿総合病院 血液内科 主任部長/副院長
萩原 政夫先生医師・歯科医師限定MDSにおけるRNAスプライシング因子・コヒーシン複合体因子の遺伝子変異
MDSにおけるRNAスプライシング因子・コヒーシン複合体因子の遺伝子変異
熊本大学大学院生命科学研究部 臨床病態解析学講座 教授/熊本大学病院 がんゲノムセンター センター長
松井 啓隆先生医師・歯科医師限定母斑症の病態と治療戦略
母斑症の病態と治療戦略
大阪大学大学院 医学系研究科 保健学専攻 神経皮膚症候群の治療法の開発と病態解析学 寄附講座教授
金田 眞理先生医師・歯科医師限定【症例紹介】薬剤性肺炎を伴ったクローン病
【症例紹介】薬剤性肺炎を伴ったクローン病
札幌医科大学医学部 消化器内科学講座/総合診療医学講座病院助教
風間 友江先生医師・歯科医師限定新たながん免疫療法の展開――治療効果を高精度に予測するバイオマーカーとは
新たながん免疫療法の展開――治療効果を高精度に予測するバイオマーカーとは
国立がん研究センター研究所 腫瘍免疫研究分野長/先端医療開発センター 免疫TR分野長
西川 博嘉先生医師・歯科医師限定AML維持療法の現状と将来性――適応患者や期待される薬剤とは
AML維持療法の現状と将来性――適応患者や期待される薬剤とは
獨協医科大学 埼玉医療センター 糖尿病内分泌・血液内科 准教授
木口 亨先生医師・歯科医師限定HIF-PH阻害薬が切り拓く新しい腎性貧血治療
HIF-PH阻害薬が切り拓く新しい腎性貧血治療
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 教授
南学 正臣先生医師・歯科医師限定糖尿病診療における遺伝学的知見――発症リスクを検出するPRSの可能性
糖尿病診療における遺伝学的知見――発症リスクを検出するPRSの可能性
琉球大学大学院医学研究科 先進ゲノム検査医学講座 教授
前田 士郎先生医師・歯科医師限定「医工連携」でチップ上に人体を再現、遠隔医療から生体モニタリングまで――国際腎臓学会が選出した60+1の「Breakthrough Discoveries:画期的な発見」から新しくも重要な「+1」
「医工連携」でチップ上に人体を再現、遠隔医療から生体モニタリングまで――国際腎臓学会が選出した60+1の「Breakthrough Discoveries:画期的な発見」から新しくも重要な「+1」
東京大学医学部附属病院 腎臓・内分泌内科 科長/教授
南学 正臣先生医師・歯科医師限定ノーベル賞につながった腎臓のHIFと低酸素症研究、腎不全との関係判明から受賞3氏が解明した低酸素症の経路――国際腎臓学会が選出した60+1の「Breakthrough Discoveries:画期的な発見」より
ノーベル賞につながった腎臓のHIFと低酸素症研究、腎不全との関係判明から受賞3氏が解明した低酸素症の経路――国際腎臓学会が選出した60+1の「Breakthrough Discoveries:画期的な発見」より
東京大学医学部附属病院 腎臓・内分泌内科 科長/教授
南学 正臣先生医師・歯科医師限定糖尿病治療薬から“大化け”したSGLT2阻害薬、腎保護作用も――国際腎臓学会選出、60+1の「Breakthrough Discoveries:画期的な発見」の1つに
糖尿病治療薬から“大化け”したSGLT2阻害薬、腎保護作用も――国際腎臓学会選出、60+1の「Breakthrough Discoveries:画期的な発見」の1つに
東京大学医学部附属病院 腎臓・内分泌内科 科長/教授
南学 正臣先生医師・歯科医師限定【症例紹介】5ASA製剤の二面性――5ASA製剤で腸炎悪化?
【症例紹介】5ASA製剤の二面性――5ASA製剤で腸炎悪化?
国立成育医療研究センター 小児内科系専門診療部 消化器科/小児炎症性腸疾患(IBD)センター
竹内 一朗先生医師・歯科医師限定2013年以降変化した喘息の検査・診断――呼気NO検査は喘息の早期発見に寄与、モストグラフは検査時の患者負担少なく
2013年以降変化した喘息の検査・診断――呼気NO検査は喘息の早期発見に寄与、モストグラフは検査時の患者負担少なく
高知大学医学部 呼吸器・アレルギー内科学教室 教授
横山 彰仁先生医師・歯科医師限定軽症喘息へのICS、LABAは定期服用から頓用へ――重症喘息とCOPDの3薬併用療法、吸入ステロイドの必要性は慎重に見極めを
軽症喘息へのICS、LABAは定期服用から頓用へ――重症喘息とCOPDの3薬併用療法、吸入ステロイドの必要性は慎重に見極めを
高知大学医学部 呼吸器・アレルギー内科学教室 教授
横山 彰仁先生医師・歯科医師限定小児喘息からCOPDへの流れ明らかに――ACO鑑別し喘息因子あればステロイド処方を
小児喘息からCOPDへの流れ明らかに――ACO鑑別し喘息因子あればステロイド処方を
高知大学医学部 呼吸器・アレルギー内科学教室 教授
横山 彰仁先生医師・歯科医師限定吸入ステロイドで激減した喘息患者の気道リモデリング――COVID-19で死亡者は2割減に
吸入ステロイドで激減した喘息患者の気道リモデリング――COVID-19で死亡者は2割減に
高知大学医学部 呼吸器・アレルギー内科学教室 教授
横山 彰仁先生医師・歯科医師限定難治性・重症喘息治療は経口ステロイドから 「生物学的製剤」へ――効果の一方、医療経済的課題も
難治性・重症喘息治療は経口ステロイドから 「生物学的製剤」へ――効果の一方、医療経済的課題も
高知大学医学部 呼吸器・アレルギー内科学教室 教授
横山 彰仁先生医師・歯科医師限定乳がんに対する免疫チェックポイント阻害薬の効果とirAE――今後の展望は
乳がんに対する免疫チェックポイント阻害薬の効果とirAE――今後の展望は
がん研有明病院 乳腺センター 副医長
尾崎 由記範先生医師・歯科医師限定HER2陽性の早期乳がんのネオアジュバント/アジュバント療法――HER2陰性化、脳転移の患者への治療戦略
HER2陽性の早期乳がんのネオアジュバント/アジュバント療法――HER2陰性化、脳転移の患者への治療戦略
東海大学医学部外科学系 乳腺内分泌外科 教授
新倉 直樹先生医師・歯科医師限定糖尿病と心不全
糖尿病と心不全
富山大学 学術研究部医学系 内科学第一 准教授
八木 邦公先生医師・歯科医師限定非メラノーマ皮膚悪性腫瘍の治療最前線――血管肉腫
非メラノーマ皮膚悪性腫瘍の治療最前線――血管肉腫
筑波大学 医学医療系 皮膚科 准教授/病院教授
藤澤 康弘先生医師・歯科医師限定コロナ禍における糖尿病に携わる医療者の役割
コロナ禍における糖尿病に携わる医療者の役割
京都府立医科大学大学院医学研究科 内分泌・代謝内科学 講師
山崎 真裕先生医師・歯科医師限定糖尿病性腎臓病(DKD)進行抑制のための包括的戦略
糖尿病性腎臓病(DKD)進行抑制のための包括的戦略
和歌山県立医科大学 腎臓内科学講座 教授
荒木 信一先生医師・歯科医師限定【論文紹介】NASH limits anti-tumour surveillance in immunotherapy-treated HCC
【論文紹介】NASH limits anti-tumour surveillance in immunotherapy-treated HCC
札幌医科大学医学部消化器内科学講座 講師
阿久津 典之先生医師・歯科医師限定【第64回日本糖尿病学会レポート】糖尿病治療薬(SGLT2阻害薬、GLP-1受容体作動薬)の腎症に対する効果とエビデンス(4700字)
【第64回日本糖尿病学会レポート】糖尿病治療薬(SGLT2阻害薬、GLP-1受容体作動薬)の腎症に対する効果とエビデンス(4700字)
内分泌系疾患>糖尿病
島根大学医学部 内科学講座 内科学第一 教授
金﨑 啓造先生医師・歯科医師限定【第120回皮膚科学会レポート】EGFR阻害薬による皮膚障害のUpdate(4200字)
【第120回皮膚科学会レポート】EGFR阻害薬による皮膚障害のUpdate(4200字)
皮膚疾患
腫瘍(オンコロジー)>その他
独立行政法人 国立病院機構 四国がんセンター皮膚科 併存症疾患センター部長
藤山 幹子先生医師・歯科医師限定【第64回日本糖尿病学会レポート】糖尿病とCOVID-19・レジストリデータ解析も含めて(4100字)
【第64回日本糖尿病学会レポート】糖尿病とCOVID-19・レジストリデータ解析も含めて(4100字)
内分泌系疾患>糖尿病
感染症>ウイルス性
国立研究開発法人 国立国際医療研究センター病院 糖尿病情報センター長
大杉 満先生医師・歯科医師限定【第120回皮膚科学会レポート】JAK阻害剤のかゆみへの効果メカニズム(4000字)
【第120回皮膚科学会レポート】JAK阻害剤のかゆみへの効果メカニズム(4000字)
皮膚疾患
免疫系疾患>アレルギー
近畿大学病院 皮膚科 医学部講師
中嶋 千紗先生医師・歯科医師限定【第64回日本糖尿病学会レポート】COVID-19の病態と治療の展望(4100字)
【第64回日本糖尿病学会レポート】COVID-19の病態と治療の展望(4100字)
感染症>ウイルス性
国立国際医療研究センター病院 国際感染症センター長
大曲 貴夫先生医師・歯科医師限定【第64回日本糖尿病学会レポート】SGLT2阻害薬 そのエビデンスの正しい読み方・使い方(4500字)
【第64回日本糖尿病学会レポート】SGLT2阻害薬 そのエビデンスの正しい読み方・使い方(4500字)
内分泌系疾患>糖尿病
聖路加国際病院 内分泌代謝科 部長
能登 洋先生医師・歯科医師限定【インタビュー】肺がん検査画像撮影の被ばく量、正しい説明で患者の不安払拭を――胸部X線はほぼノーリスク、精密検査用CTは5回でがんリスク0.5%上昇(700字))
【インタビュー】肺がん検査画像撮影の被ばく量、正しい説明で患者の不安払拭を――胸部X線はほぼノーリスク、精密検査用CTは5回でがんリスク0.5%上昇(700字))
その他>検査
日本大学医学部附属 板橋病院 呼吸器外科 部長、日本大学 医学部 外科学系 呼吸器外科学分野 主任教授
櫻井 裕幸先生医師・歯科医師限定【症例紹介】潰瘍性大腸炎合併大腸がんと治療について(2500字)
【症例紹介】潰瘍性大腸炎合併大腸がんと治療について(2500字)
腫瘍(オンコロジー)>消化器
消化器疾患>消化管
札幌医科大学 医学部 消化器内科学講座 診察医
林 優希先生医師・歯科医師限定【第120回皮膚科学会レポート】脊髄を切り口とした慢性掻痒症の新しいメカニズム(5600字)
【第120回皮膚科学会レポート】脊髄を切り口とした慢性掻痒症の新しいメカニズム(5600字)
皮膚疾患
九州大学 大学院薬学研究院 薬理学分野 助教
白鳥 美穂先生医師・歯科医師限定【インタビュー】心不全増加の主要因は高齢化、ほかには先天性心疾患、がんの治療の進歩――“治す”には根本的原因の究明が不可欠(1100字)
【インタビュー】心不全増加の主要因は高齢化、ほかには先天性心疾患、がんの治療の進歩――“治す”には根本的原因の究明が不可欠(1100字)
心臓血管疾患>心臓
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】心不全治療20年ぶり新薬4種――最注目はSGLT2阻害薬、世界で初めてHFpEFにも有効性を発揮(1100字)
【インタビュー】心不全治療20年ぶり新薬4種――最注目はSGLT2阻害薬、世界で初めてHFpEFにも有効性を発揮(1100字)
心臓血管疾患>心臓
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】実は多かった心アミロイドーシス患者、診断にピロリン酸シンチが有効――タファミジスで総死亡・心血管疾患による入院を抑制(600字)
【インタビュー】実は多かった心アミロイドーシス患者、診断にピロリン酸シンチが有効――タファミジスで総死亡・心血管疾患による入院を抑制(600字)
心臓血管疾患>心臓
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】心不全予防のチャンスは4回――正しい知識の周知と予防で死亡回避を(900字)
【インタビュー】心不全予防のチャンスは4回――正しい知識の周知と予防で死亡回避を(900字)
心臓血管疾患>心臓
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】非細菌・非ウイルス性炎症に新概念「クローナル・ヘマトポイエーシス」――経口薬で抑制の可能性に現実味(780字)
【インタビュー】非細菌・非ウイルス性炎症に新概念「クローナル・ヘマトポイエーシス」――経口薬で抑制の可能性に現実味(780字)
その他>その他
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】新型コロナでも多い心不全死――ワクチンでの発症なら軽微、接種控えの理由にならず(600字)
【インタビュー】新型コロナでも多い心不全死――ワクチンでの発症なら軽微、接種控えの理由にならず(600字)
感染症>ウイルス性
その他>ワクチン
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】AIやウエアラブルデバイスでの心房細動早期発見に期待――脳卒中予防の強力ツールに(800字)
【インタビュー】AIやウエアラブルデバイスでの心房細動早期発見に期待――脳卒中予防の強力ツールに(800字)
心臓血管疾患>心臓
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】がんの適応広がる免疫チェックポイント阻害薬、心筋炎の原因にも――腫瘍循環器分野の基礎研究進展を(600字)
【インタビュー】がんの適応広がる免疫チェックポイント阻害薬、心筋炎の原因にも――腫瘍循環器分野の基礎研究進展を(600字)
その他>薬物治療
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【インタビュー】密接になるがんと心疾患――腫瘍と循環器の専門医 連携が必要(700字)
【インタビュー】密接になるがんと心疾患――腫瘍と循環器の専門医 連携が必要(700字)
その他>薬物治療
東京大学大学院医学系研究科 内科学専攻器官病態内科学講座 循環器内科学教授
小室 一成先生医師・歯科医師限定【第120回皮膚科学会レポート】湿疹三角を読み解く(2200字)
【第120回皮膚科学会レポート】湿疹三角を読み解く(2200字)
皮膚疾患
中東遠総合医療センター 参与、皮膚科・皮膚腫瘍科診療部長、アレルギー疾患研究センター長
戸倉 新樹先生医師・歯科医師限定【論文紹介】Induced organoids derived from patients with ulcerative colitis recapitulate colitic reactivity(1700字)
【論文紹介】Induced organoids derived from patients with ulcerative colitis recapitulate colitic reactivity(1700字)
消化器疾患>消化管
札幌医科大学医学部 消化器内科学講座 特任助教
平山 大輔先生医師・歯科医師限定【第120回皮膚科学会レポート】全身炎症と乾癬(4700字)
【第120回皮膚科学会レポート】全身炎症と乾癬(4700字)
帝京大学医学部 皮膚科学講座 主任教授
多田 弥生先生医師・歯科医師限定【第120回皮膚科学会レポート】好酸球性筋膜炎・硬化性萎縮性苔癬の診断と治療(5400字)
【第120回皮膚科学会レポート】好酸球性筋膜炎・硬化性萎縮性苔癬の診断と治療(5400字)
皮膚疾患
福井大学医学部 皮膚科学講座
宇都宮 慧先生医師・歯科医師限定【症例紹介】深部静脈血栓・門脈血栓・Trousseau症候群を併発した膵体尾部がんの一例(1500字)
【症例紹介】深部静脈血栓・門脈血栓・Trousseau症候群を併発した膵体尾部がんの一例(1500字)
腫瘍(オンコロジー)>消化器
札幌医科大学医学部 消化器内科学講座 助教
石上 敬介先生医師・歯科医師限定【症例紹介】家族性大腸ポリポーシスに合併したステージIV大腸がんの治療(1600字)
【症例紹介】家族性大腸ポリポーシスに合併したステージIV大腸がんの治療(1600字)
腫瘍(オンコロジー)>消化器
札幌医科大学医学部 消化器内科学講座
大和田 紗恵先生医師・歯科医師限定【第120回皮膚科学会レポート】新しい薬剤誘発性type-1過敏症(2700字)
【第120回皮膚科学会レポート】新しい薬剤誘発性type-1過敏症(2700字)
皮膚疾患
磐田市立総合病院 皮膚科 部長
橋爪 秀夫先生医師・歯科医師限定【インタビュー】心臓移植「不適応」患者にも植込型補助人工心臓の適応を拡大――管理可能な施設など体制整備を推進(540字)
【インタビュー】心臓移植「不適応」患者にも植込型補助人工心臓の適応を拡大――管理可能な施設など体制整備を推進(540字)
心臓血管疾患>心臓
日本胸部外科学会統括会長 慶應義塾大学 医学部外科学 教授
志水 秀行先生医師・歯科医師限定【症例紹介】Epstein-Barr virus(EBV)とチオプリン製剤投与――EBV感染状況の確認(1200字)
【症例紹介】Epstein-Barr virus(EBV)とチオプリン製剤投与――EBV感染状況の確認(1200字)
消化器疾患>消化管
免疫系疾患>自己免疫疾患
感染症>ウイルス性
札幌医科大学医学部 消化器内科学講座
横山 佳浩先生医師・歯科医師限定【論文紹介】Short-term Outcomes of Robotic Gastrectomy vs Laparoscopic Gastrectomy for Patients With Gastric Cancer: A Randomized Clinical Trial(2000字)
【論文紹介】Short-term Outcomes of Robotic Gastrectomy vs Laparoscopic Gastrectomy for Patients With Gastric Cancer: A Randomized Clinical Trial(2000字)
腫瘍(オンコロジー)>消化器
和歌山県立医科大学 外科学第二講座 講師
尾島 敏康先生医師・歯科医師限定【第120回皮膚科学会レポート】新しい皮膚そう痒症ガイドラインの概要(3900字)
【第120回皮膚科学会レポート】新しい皮膚そう痒症ガイドラインの概要(3900字)
皮膚疾患
防衛医科大学校 皮膚科学講座 教授
佐藤 貴浩先生医師・歯科医師限定【症例紹介】微小大腸がんの1例:拡大内視鏡観察の重要性(1100字)
【症例紹介】微小大腸がんの1例:拡大内視鏡観察の重要性(1100字)
腫瘍(オンコロジー)>消化器
札幌医科大学医学部 消化器内科学講座
吉井新二先生医師・歯科医師限定【インタビュー】「形、機能、命を守る」テーマに診療科・臓器横断的な議論展開――日本癌治療学会学術集会10月末に横浜で開催(2400字)
【インタビュー】「形、機能、命を守る」テーマに診療科・臓器横断的な議論展開――日本癌治療学会学術集会10月末に横浜で開催(2400字)
腫瘍(オンコロジー)>その他
国立がん研究センター東病院 副院長
林 隆一先生医師・歯科医師限定【インタビュー】転移性尿路上皮がんに対する新たな治療薬「エンホルツマブ ベドチン」(490字)
【インタビュー】転移性尿路上皮がんに対する新たな治療薬「エンホルツマブ ベドチン」(490字)
腫瘍(オンコロジー)>泌尿生殖器
獨協医科大学埼玉医療センター 泌尿器科教授/低侵襲治療センター長
井手 久満先生医師・歯科医師限定【インタビュー】膀胱がん手術時の光線力学的診断――目視できないがん細胞判別も可能に(370字)
【インタビュー】膀胱がん手術時の光線力学的診断――目視できないがん細胞判別も可能に(370字)
腫瘍(オンコロジー)>泌尿生殖器
獨協医科大学埼玉医療センター 泌尿器科教授/低侵襲治療センター長
井手 久満先生医師・歯科医師限定【インタビュー】健康状態・寿命との相関にも注目集まるテストステロン――爪測定法の開発に向けて(570字)
【インタビュー】健康状態・寿命との相関にも注目集まるテストステロン――爪測定法の開発に向けて(570字)
泌尿生殖器疾患>男性
内分泌系疾患>その他
獨協医科大学埼玉医療センター 泌尿器科教授/低侵襲治療センター長
井手 久満先生医師・歯科医師限定【第120回皮膚科学会レポート】蕁麻疹に対するオマリズマブ治療(2800字)
【第120回皮膚科学会レポート】蕁麻疹に対するオマリズマブ治療(2800字)
皮膚疾患
広島大学大学院 皮膚科学 准教授
田中 暁生先生医師・歯科医師限定【インタビュー】胃癌治療ガイドライン2021年7月改訂の要点――未分化型粘膜内がんがESD適応に(780字)
【インタビュー】胃癌治療ガイドライン2021年7月改訂の要点――未分化型粘膜内がんがESD適応に(780字)
腫瘍(オンコロジー)>消化器
東京大学大学院医学系研究科 消化器内科学 教授
藤城 光弘先生医師・歯科医師限定【インタビュー】胃癌治療ガイドライン2021年7月改訂の要点――高齢者に対する内視鏡的切除は推奨されるか(670字)
【インタビュー】胃癌治療ガイドライン2021年7月改訂の要点――高齢者に対する内視鏡的切除は推奨されるか(670字)
腫瘍(オンコロジー)>消化器
東京大学大学院医学系研究科 消化器内科学 教授
藤城 光弘先生医師・歯科医師限定【インタビュー】胃癌治療ガイドライン2021年7月改訂の要点――抗血栓薬服用者に対する内視鏡的切除は推奨されるか(650字)
【インタビュー】胃癌治療ガイドライン2021年7月改訂の要点――抗血栓薬服用者に対する内視鏡的切除は推奨されるか(650字)
腫瘍(オンコロジー)>消化器
東京大学大学院医学系研究科 消化器内科学 教授
藤城 光弘先生医師・歯科医師限定【インタビュー】早期胃がんに対するESD後の出血リスクを予測する「BEST-Jスコア」とは(460字)
【インタビュー】早期胃がんに対するESD後の出血リスクを予測する「BEST-Jスコア」とは(460字)
腫瘍(オンコロジー)>消化器
東京大学大学院医学系研究科 消化器内科学 教授
藤城 光弘先生医師・歯科医師限定【インタビュー】食道がんに対する内視鏡的切除後の狭窄をいかに予防するか――臨床研究の進展(480字)
【インタビュー】食道がんに対する内視鏡的切除後の狭窄をいかに予防するか――臨床研究の進展(480字)
腫瘍(オンコロジー)>消化器
東京大学大学院医学系研究科 消化器内科学 教授
藤城 光弘先生医師・歯科医師限定【インタビュー】「貯筋」につながるレジスタンス運動――超高齢社会で高まる重要性(1900字)
【インタビュー】「貯筋」につながるレジスタンス運動――超高齢社会で高まる重要性(1900字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】低血糖を起こさずにインスリン分泌能を保つDPP-4阻害薬(1400字)
【インタビュー】低血糖を起こさずにインスリン分泌能を保つDPP-4阻害薬(1400字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】インスリン抵抗性を改善するビグアナイド薬とチアゾリジン薬(610字)
【インタビュー】インスリン抵抗性を改善するビグアナイド薬とチアゾリジン薬(610字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】多面的な作用をもたらすSGLT2阻害薬活用の可能性とは(1300字)
【インタビュー】多面的な作用をもたらすSGLT2阻害薬活用の可能性とは(1300字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】SGLT2阻害薬の意外な作用――心不全を抑制する可能性も(550字)
【インタビュー】SGLT2阻害薬の意外な作用――心不全を抑制する可能性も(550字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】GLP-1受容体作動薬の進歩――週1回の自己注射薬と使い分けの考え方(780字)
【インタビュー】GLP-1受容体作動薬の進歩――週1回の自己注射薬と使い分けの考え方(780字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】GLP-1受容体作動薬に経口薬が登場――「痩せ薬」としての使用に警鐘も(720字)
【インタビュー】GLP-1受容体作動薬に経口薬が登場――「痩せ薬」としての使用に警鐘も(720字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】糖尿病「第4の治療」、肥満外科手術療法のメリットと安全性(820字)
【インタビュー】糖尿病「第4の治療」、肥満外科手術療法のメリットと安全性(820字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】糖尿病と精密医療――日本人特有の遺伝子の発見、今後の可能性(910字)
【インタビュー】糖尿病と精密医療――日本人特有の遺伝子の発見、今後の可能性(910字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【インタビュー】精密医療による糖尿病合併症の解明――遺伝子情報による差別是正の必要も(1100字)
【インタビュー】精密医療による糖尿病合併症の解明――遺伝子情報による差別是正の必要も(1100字)
内分泌系疾患>糖尿病
虎の門病院 院長
門脇 孝先生医師・歯科医師限定【第120回皮膚科学会レポート】皮膚と多臓器病変をつなぐ:全身性強皮症(2600字)
【第120回皮膚科学会レポート】皮膚と多臓器病変をつなぐ:全身性強皮症(2600字)
皮膚疾患
東京大学大学院医学系研究科・医学部 皮膚科准教授
浅野 善英先生医師・歯科医師限定【第120回皮膚科学会レポート】痒みに対するプラセボ・ノセボ効果(4500字)
【第120回皮膚科学会レポート】痒みに対するプラセボ・ノセボ効果(4500字)
皮膚疾患
東京慈恵会医科大学皮膚科学講座 講師
石氏 陽三先生医師・歯科医師限定【インタビュー】治療選択のパラダイムシフト――切除不能胃がんの1次治療に免疫療法も(780字)
【インタビュー】治療選択のパラダイムシフト――切除不能胃がんの1次治療に免疫療法も(780字)
腫瘍(オンコロジー)>消化器
名古屋大学大学院医学系研究科 消化器外科学講座 教授
小寺 泰弘先生医師・歯科医師限定【インタビュー】HER2陽性胃がんに対する抗がん剤「トラスツズマブ デルクステカン」の承認(460字)
【インタビュー】HER2陽性胃がんに対する抗がん剤「トラスツズマブ デルクステカン」の承認(460字)
腫瘍(オンコロジー)>消化器
名古屋大学大学院医学系研究科 消化器外科学講座 教授
小寺 泰弘先生医師・歯科医師限定【インタビュー】手術療法の進歩――消化器外科領域のロボット支援下手術、課題と可能性は(1300字)
【インタビュー】手術療法の進歩――消化器外科領域のロボット支援下手術、課題と可能性は(1300字)
消化器疾患>その他
名古屋大学大学院医学系研究科 消化器外科学講座 教授
小寺 泰弘先生医師・歯科医師限定【インタビュー】薬物療法の進歩によりコンバージョン手術できる症例が増加(350字)
【インタビュー】薬物療法の進歩によりコンバージョン手術できる症例が増加(350字)
腫瘍(オンコロジー)>消化器
名古屋大学大学院医学系研究科 消化器外科学講座 教授
小寺 泰弘先生医師・歯科医師限定【インタビュー】腹膜播種に対する治療の進歩と課題――テガフール・ギメラシル・オテラシルカリウム配合剤やパクリタキセルの活用は(480字)
【インタビュー】腹膜播種に対する治療の進歩と課題――テガフール・ギメラシル・オテラシルカリウム配合剤やパクリタキセルの活用は(480字)
腫瘍(オンコロジー)>消化器
名古屋大学大学院医学系研究科 消化器外科学講座 教授
小寺 泰弘先生医師・歯科医師限定【インタビュー】ペイシェント・アドボカシーの進展――胃がん領域には課題も(560字)
【インタビュー】ペイシェント・アドボカシーの進展――胃がん領域には課題も(560字)
腫瘍(オンコロジー)>消化器
名古屋大学大学院医学系研究科 消化器外科学講座 教授
小寺 泰弘先生医師・歯科医師限定【第120回皮膚科学会レポート】急速進行性間質性肺疾患の早期診断に重要な皮膚所見(3100字)
【第120回皮膚科学会レポート】急速進行性間質性肺疾患の早期診断に重要な皮膚所見(3100字)
皮膚疾患
大阪大学大学院医学系研究科 皮膚科学教室 特任講師
植田 郁子先生医師・歯科医師限定【インタビュー】免疫細胞と腫瘍細胞を“強制結合”――CAR-Tに続くBiTE抗体薬に注目(240字)
【インタビュー】免疫細胞と腫瘍細胞を“強制結合”――CAR-Tに続くBiTE抗体薬に注目(240字)
腫瘍(オンコロジー)>その他
東北大学大学院医学系研究科・医学部 血液・免疫病学分野 教授
張替 秀郎先生医師・歯科医師限定【インタビュー】分子標的薬の開発は多様性、速度もアップ――臨床医も作用機序理解に分子生物学的知識の更新を(630字)
【インタビュー】分子標的薬の開発は多様性、速度もアップ――臨床医も作用機序理解に分子生物学的知識の更新を(630字)
腫瘍(オンコロジー)>その他
東北大学大学院医学系研究科・医学部 血液・免疫病学分野 教授
張替 秀郎先生医師・歯科医師限定【インタビュー】がん細胞の「不均一性」は時空間的――シングルセル解析で個々の性質を把握し、より精密な研究が可能に(750字)
【インタビュー】がん細胞の「不均一性」は時空間的――シングルセル解析で個々の性質を把握し、より精密な研究が可能に(750字)
腫瘍(オンコロジー)>その他
慶應義塾大学医学部 先端医科学研究所 遺伝子制御研究部門 教授
佐谷 秀行先生医師・歯科医師限定【インタビュー】新型コロナワクチンで開発が加速――がんワクチンはRNAが主役に(350字)
【インタビュー】新型コロナワクチンで開発が加速――がんワクチンはRNAが主役に(350字)
その他>ワクチン
慶應義塾大学医学部 先端医科学研究所 遺伝子制御研究部門 教授
佐谷 秀行先生医師・歯科医師限定【インタビュー】80年の歴史回顧し未来を展望――日本癌学会学術総会9月末から横浜で(3300字)
【インタビュー】80年の歴史回顧し未来を展望――日本癌学会学術総会9月末から横浜で(3300字)
腫瘍(オンコロジー)>その他
慶應義塾大学医学部 先端医科学研究所 遺伝子制御研究部門 教授
佐谷 秀行先生医師・歯科医師限定【インタビュー】欧米を中心に進む化学療法+放射線治療による腫瘍縮小――ストーマ造設回避も(410字)
【インタビュー】欧米を中心に進む化学療法+放射線治療による腫瘍縮小――ストーマ造設回避も(410字)
腫瘍(オンコロジー)>消化器
尼崎中央病院 副院長/消化器病センター長
松原 長秀先生医師・歯科医師限定【インタビュー】全ゲノム解析が変えるがん治療の可能性――「二次的所見」取り扱いには配慮必要(630字)
【インタビュー】全ゲノム解析が変えるがん治療の可能性――「二次的所見」取り扱いには配慮必要(630字)
腫瘍(オンコロジー)>その他
尼崎中央病院 副院長/消化器病センター長
松原 長秀先生医師・歯科医師限定【インタビュー】免疫チェックポイント阻害薬が遺伝性大腸がんの一部に特異的効果――ワクチン療法にも期待(480字)
【インタビュー】免疫チェックポイント阻害薬が遺伝性大腸がんの一部に特異的効果――ワクチン療法にも期待(480字)
腫瘍(オンコロジー)>消化器
腫瘍(オンコロジー)>その他
尼崎中央病院 副院長/消化器病センター長
松原 長秀先生医師・歯科医師限定【インタビュー】術前化学療法が効かなかった場合に術後補助療法で予後が改善――トリプルネガティブにはカペシタビン、HER2陽性には新たなHER2阻害薬で(530字)
【インタビュー】術前化学療法が効かなかった場合に術後補助療法で予後が改善――トリプルネガティブにはカペシタビン、HER2陽性には新たなHER2阻害薬で(530字)
腫瘍(オンコロジー)>乳腺・乳房
東京医科大学病院乳腺科主任教授/日本乳癌学会理事
石川 孝先生医師・歯科医師限定【インタビュー】薬剤への反応から「次の方法」を考えるレスポンスガイド――ホルモン受容体陰性がんでは実臨床に(460字)
【インタビュー】薬剤への反応から「次の方法」を考えるレスポンスガイド――ホルモン受容体陰性がんでは実臨床に(460字)
腫瘍(オンコロジー)>乳腺・乳房
東京医科大学病院乳腺科主任教授/日本乳癌学会理事
石川 孝先生医師・歯科医師限定【インタビュー】乳がん治療にも免疫チェックポイント阻害剤導入始まる――対象を選ぶ必要も(280字)
【インタビュー】乳がん治療にも免疫チェックポイント阻害剤導入始まる――対象を選ぶ必要も(280字)
腫瘍(オンコロジー)>乳腺・乳房
東京医科大学病院乳腺科主任教授/日本乳癌学会理事
石川 孝先生医師・歯科医師限定【インタビュー】遺伝性乳がんなどBRCA遺伝子変異にはPARP阻害剤が著効――卵巣がんなどへの臓器横断的治療も視野に(680字)
【インタビュー】遺伝性乳がんなどBRCA遺伝子変異にはPARP阻害剤が著効――卵巣がんなどへの臓器横断的治療も視野に(680字)
腫瘍(オンコロジー)>乳腺・乳房
東京医科大学病院乳腺科主任教授/日本乳癌学会理事
石川 孝先生医師・歯科医師限定【インタビュー】遺伝子パネル診断は早期導入でデータ収集が必要――繰り返し検体採取で解決へ(350字)
【インタビュー】遺伝子パネル診断は早期導入でデータ収集が必要――繰り返し検体採取で解決へ(350字)
腫瘍(オンコロジー)>乳腺・乳房
東京医科大学病院乳腺科主任教授/日本乳癌学会理事
石川 孝先生医師・歯科医師限定【インタビュー】エビデンスの「海外直輸入」には一考の余地――体型差や薬剤感受性の違いなどで異なる結果になることも(510字)
【インタビュー】エビデンスの「海外直輸入」には一考の余地――体型差や薬剤感受性の違いなどで異なる結果になることも(510字)
腫瘍(オンコロジー)>乳腺・乳房
東京医科大学病院乳腺科主任教授/日本乳癌学会理事
石川 孝先生医師・歯科医師限定【第64回糖尿病学会レポート】糖尿病とがん、そして腫瘍糖尿病学へ(5700字)
【第64回糖尿病学会レポート】糖尿病とがん、そして腫瘍糖尿病学へ(5700字)
腫瘍(オンコロジー)>その他
内分泌系疾患>糖尿病
国立がんセンター中央病院総合内科(糖尿病腫瘍科)
大橋 健先生医師・歯科医師限定【第120回皮膚科学会レポート】皮膚からアプローチする腫瘍免疫(3400字)
【第120回皮膚科学会レポート】皮膚からアプローチする腫瘍免疫(3400字)
腫瘍(オンコロジー)>皮膚
近畿大学医学部皮膚科学教室 主任教授
大塚 篤司先生医師・歯科医師限定【第64回糖尿病学会レポート】糖尿病と心不全の新しい関係(3300字)
【第64回糖尿病学会レポート】糖尿病と心不全の新しい関係(3300字)
心臓血管疾患>心臓
内分泌系疾患>糖尿病
富山大学大学院医学薬学研究部内科学第二(第二内科)教授
絹川 弘一郎先生医師・歯科医師限定【第120回皮膚科学会レポート】免疫チェックポイント阻害薬による皮膚障害 Update(3400字)
【第120回皮膚科学会レポート】免疫チェックポイント阻害薬による皮膚障害 Update(3400字)
皮膚疾患
腫瘍(オンコロジー)>その他
その他>薬物治療
横浜市立大学大学院医学研究科 環境免疫病態皮膚科学 教授
山口 由衣先生医師・歯科医師限定【論文紹介】Cell-Free Virus-Host Chimera DNA From Hepatitis B Virus Integration Sites as a Circulating Biomarker of Hepatocellular Cancer(1600字)
【論文紹介】Cell-Free Virus-Host Chimera DNA From Hepatitis B Virus Integration Sites as a Circulating Biomarker of Hepatocellular Cancer(1600字)
腫瘍(オンコロジー)>消化器
その他>検査
スタンフォード大学医学部 微生物学・免疫学教室
關場 一磨先生医師・歯科医師限定